Download

1 / 1
10 likes | 102 Views
Apply an exact Bayesian network learning algorithm to reveal regulatory relationships among epigenetic features in human CD4 cells. Learn about the epigenetic code, chromatin structure, and optimal learning techniques for ChIP-Seq data analysis.

E N D
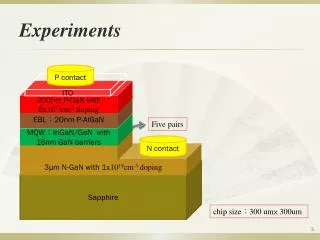
Seq-ing the Epigentic Code with Exact Bayesian Network Structure Learning Brandon M. Malone1,2, Changhe Yuan1, Eric Hansen1 and Susan M. Bridges1,2 1Department of Computer Science & Engineering, Mississippi State University 2Institute for Genomics, Biocomputing and Biotechnology, Mississippi State University . 1,2,3,4 H3K9 ac H3K9 ac H3K9 ac H3K9 ac H3K4 me3 H3K4 me3 H3K4 me3 H3K4 me3 Expand(U) For each X in U newScore = U.score + BestScore(U, X) succ = get({U+ X}) if newScore < succ.score put({U+ X}, newScore) 1,2,3 1,2,4 ϕ 1,3,4 2,3,4 A B 1,2 1,3 2,3 1,4 2,4 3,4 Pol II Pol II Pol II Pol II H3K27me3 H3K27me3 H3K27me3 H3K27me3 a a b b Vary NodeNx,u AD Node Nu B B 1 2 3 4 Abstract • The epigenetic code [Jaenisch] hypothesis proposes that patterns of post-translational modifications to the histone core proteins, the presence of transcription factor binding sites and other genomic features influence expression of associated DNA. Chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-Seq) is frequently used to characterize these features at a genome-wide scale. Previous studies [Yu] have used approximation techniques to learn relationships among them. In this work, we apply a novel exact Bayesian network learning algorithm to learn a network structure which identifies regulatory relationships among a set of epigenetic features in human CD4 cells [Barksi]. Comparison to networks learned using greedy methods reveals that our network identifies more biologically relevant relationships. By applying an exact, optimal learning algorithm instead of an approximate, greedy algorithm, the relationships we learn are unaffected by sources of uncertainty stemming from the structure learning algorithm. H3K36me H3K36me H3K36me Expr ab ab ab ab ϕ Optimal Learning with Dynamic Programming In the case of a ChIP-Seq dataset, we do not know the relationships among the variables. Therefore, we must learn them. Singh and Moore [2005] proposed a dynamic programming algorithm to learn an optimal Bayesian network which minimizes the MDL score. The figure below shows the intuition behind the algorithm. Equation 2 expresses this recursively. Silander and Myllmaki [2006] refined the algorithm by reversing the process. • The Epigenetic Code • The central dogma of molecular biology (roughly) states that DNA is transcribed into RNA which is translated into proteins. Proteins perform many of the functions in the body. We have the same DNA in most of our cells, yet they perform quite different functions. One reason for this differentiation lies in the epigenetic code.When DNA forms chromosomes, it packs together very tightly into a structure called chromatin. The DNA coils around a group of eight proteins called histones. Figure 1 summarizes chromatin packaging.The histone proteins include a tail domain which is very susceptible to a large number of post-translational modifications which affect the attraction between histones. The attraction can increase between histones, tightening surrounding chromatin and suppressing expression. Chromatin can also loosen, increasing expression.The combination of present modifications determines the effect on the chromatin structure. Some histone modifications affect the likelihood of other modifications. The epigenetic code [Jaenisch] proposes that the combination of histone modifications, as well as other features such as the presence of transcription factor binding sites, serves as a type of message to present and future generations of cells about regulation. Figure 1. Chromatin packaging and histones.(http://themedicalbiochemistrypage.org/) Equations (1) (2) Remove that leaf and its edges from the network.. The remaining subnetwork is also a DAG, so it has a leaf. Recursively find optimal leaves until an empty subnetwork remains. The optimal Bayesian network structure is a DAG, so it has a leaf variable with no children. • Our Optimal Search Formulation • As suggested by Equation 2, learning an optimal Bayesian network consists of three phases which we formulate as search problems. Calculating Scores Goal. Calculate MDL(X|U), which is the score of X using U as parents Representation. AD-tree [Moore] Search Strategy. Depth-first AD Node. Records with U= u Vary Node. Records with U = u,X = x Successor. Instantiate a new X Storage. Written to disk Identifying Optimal Parent Sets Goal. Calculate BestScore(U, X), whichselects the best parents of X from U Representation. Sorted and bit arrays Search Strategy. On demand Successor. Use bit operators to find scores consistent with U\Y Score. scores[firstBit(usable(X))] Storage. Arrays and bit sets Learning Optimal Subnetworks Goal. Calculate Score(U), which is the best subnetwork for variables U. Representation. Order graph [Yuan] Search Strategy. Breadth-first Node. Score(U) for some U. Successor. Use X as a leaf of U Score. Score(U) + BestScore(U, X) Storage. Hash table or written to disk • ChIP-Seq • We can measure the presence of a particular histone modification in cells using chromatin immunopreciptation followed by high throughput sequencing (ChIP-Seq). The figure below shows the ChIP-Seq process. Calculate and sort all of the scores for a variable. Mark which scores use each variable (n-1 of these each). Initially, a variable can use all scores. The first is optimal. When X is used as a leaf, find the usable parent scores with (usable & ~uses[X]). The first set bit is optimal. Raw DNA The DNA is sheared into pieces around 200 bp in length. Frontier Breadth-first Branch and Bound Search The order graph has a very regular structure. The successors for a node in layer l always appear in layer l+1. This observation allows us to keep only two layers in memory rather than all n. Furthermore, we can calculate how good a particular node can possibly be. If this is worse than a known bound, we safely disregard it. If optimality is not needed, we disregard many nodes to reduce running time. Pieces are immunoprecipitatedagainst an antibody to extract desired pieces. The remaining pieces of DNA are sequenced. Experiments We used our optimal frontier breadth-first search algorithm to learn an optimal Bayesian network over the 23-variable data set and compared it to a greedy search, used previously [Yu]. Figures 2 and 3 show the learned networks. • Bayesian Networks • Representation. Joint probability distribution over a set of variables • Structure. Directed acyclic graph storing conditional dependencies. • Vertices correspond to variables. • Edges indicate relationships among variables. • Parameters. Conditional probability tables quantifying relationships • Scoring. Minimum Description Length (MDL) [Schwartz], Equation 1 The sequenced DNA is mapped back to the genome. [Illumina] Results and Discussion We focused on the transcription factor binding site for CTCF, known to play a function in the regulation of many elements. We expect CTCF to be an ancestor of important regulatory elements. In our network, CTCF is parent of the five most highly connected regulatory elements in the network. The approximate algorithm identified four parents and three children of intermediate degree for CTCF. Data Set and Preprocessing Raw Data. 30 human ChIP-Seq experiments [Barski] Cellular Environment. CD4 cells (specialized white blood cells)Normalization. Linear regression, against an IgG control data setDiscretization. Clustered genes using MDL for each experiment Processed Data Set. A numeric array of length 30 for each gene Figure 2. Learned structure with our optimal algorithm. Figure 3. Learned structure with a standard greedy algorithm. Selected References • Jaenisch, R. & Bird, A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals Nature Genetics, 2003, 33, 245-254Schwarz, G. (1978). "Estimating the Dimension of a Model." The Annals of Statistics 6(2): 461-464.Barski, A., S. Cuddapah, et al. (2007). "High-resolution profiling of histonemethylations in the human genome." Cell 129(4): 823 – 837 • Singh, A. P. and A. W. Moore (2005). Finding optimal bayesian networks by dynamic programming (Technical Report). Carnegie Mellon Univ: 05—106.Silander, T. and P. Myllymaki (2006). A simple approach for finding the globally optimal Bayesian network structure. Proceedings of the 22nd Annual Conference on Uncertainty in Artificial Intelligence (UAI-06), AUAI Press. • Yu, H., S. Zhu, et al. (2008). "Inferring causal relationships among different histone modifications and gene expression." Genome Research 18(8): 1314-1324. • Yuan, C.; Malone, B. & Wu, X. Learning Optimal Bayesian Networks using A* Search. Proceedings of the 22nd International Joint Conference on Artificial Intelligence, 2011 Conclusions We presented a frontier breadth-first search algorithm for learning optimal Bayesian networks that improves the memory complexity from O(2n) to O(C(n,n/2)). Provably optimal solutions allow us to focus on interpreting the results. We learned the optimal structure of a network of epigeneitc features; it included more biologically meaningful relationships than structures learned with greedy search. Acknowledgments This material is based on work supported by the National Science Foundation under Grants No. NSF EPS-0903787 and NSF IIS-0953723.