Download

1 / 43
430 likes | 558 Views
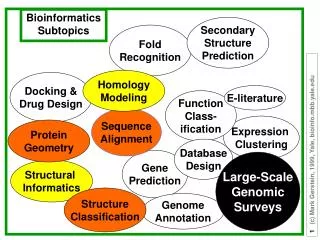
Protein Fold recognition. Morten Nielsen, CBS, BioCentrum, DTU. Outline. Many textbooks and experts state that %ID is the only determining factor for successful homology modeling This is WRONG! %ID is a very poor measure to determine if a protein can be modeled

E N D
Protein Fold recognition Morten Nielsen, CBS, BioCentrum, DTU
Outline • Many textbooks and experts state that %ID is the only determining factor for successful homology modeling • This is WRONG! • %ID is a very poor measure to determine if a protein can be modeled • Many sequences with sequence homology ~10-15% can be accurately modeled
Outline • Why homology modeling • How is it done • How to decide when to use homology modeling • Why is %id such a terrible measure • What are the best methods
Why protein modeling? • Because it works! • Close to 50% of all new sequences can be homology modeled • Experimental effort to determine protein structure is very large and costly • The gap between the size of the protein sequence data and protein structure data is large and increasing
Homology modeling and the human genome Human genome ~ 30.000 proteins
~200.000 in Swiss-Prot ~ 2.000.000 if include Tremble Swiss-Prot database
PDB New Fold Growth • The number of unique folds in nature is fairly small (possibly a few thousands) • 90% of new structures submitted to PDB in the past three years have similar structural folds in PDB Old folds New PDB structures New folds
Identification of fold • If sequence similarity is high proteins share structure (Safe zone) • If sequence similarity is low proteins may share structure (Twilight zone) • Most proteins do not have a high sequence homologous partner Rajesh Nair & Burkhard Rost Protein Science, 2002, 11, 2836-47
Why %id is so bad!! 1200 models sharing 25-95% sequence identity with the submitted sequences (www.expasy.ch/swissmod)
Identification of correct fold • % ID is a poor measure • Many evolutionary related proteins share low sequence homology • Alignment score even worse • Many sequences will score high against every thing (hydrophobic stretches) • P-value or E-value more reliable
Score 150 10 hits with higher score (E=10) 10000 hits in database => P=10/10000 = 0.001 P(Score) Score What are P and E values? • E-value • Number of expected hits in database with score higher than match • Depends on database size • P-value • Probability that a random hit will have score higher than match • Database size independent
Identify fold (template) for modeling Find the structure in the PDB database that resembles your new protein the most Can be used to predict function Align protein sequence to template Simple alignment methods Sequence profiles Threading methods Pseudo force fields Model side chains and loops How to do it
Protein superfamily Protein world New Fold Protein family Protein fold Protein structure classification
Superfamilies • Proteins which are (remote) evolutionarily related • Sequence similarity low • Share function • Share special structural features • Relationships between members of a superfamily may not be readily recognizable from the sequence alone Fold Superfamily Family Proteins
Template identification • Simple sequence based methods • Align (BLAST) sequence against sequence of proteins with known structure (PDB database) • Sequence profile based methods • Align sequence profile (Psi-BLAST) against sequence of proteins with known structure (PDB) • Align sequence profile against profile of proteins with known structure (FFAS) • Sequence and structure based methods • Align profile and predicted secondary structure against proteins with known structure (3D-PSSM)
Sequence profiles • In conventional alignment, a scoring matrix (BLOSUM62) gives the score for matching two amino acids • In reality not all positions in a protein are equally likely to mutate • Some amino acids (active cites) are highly conserved, and the score for mismatch must be very high • Other amino acids can mutate almost for free, and the score for mismatch is lower than the BLOSUM score • Sequence profiles (just like a HMM) can capture these differences
Sequence profiles TVNGQ--FPGPRLAGVAREGDQVLVKVVNHVAENITIHWHGVQLGTGWADGPAYVTQCPI TKAVVLTFNTSVEICLVMQGTSIVAAESHPLHLHGFNFPSNFNLVDPMERNTAGVP a)TKAVVLTFNTSVEICLVMQGTSIV----AAESHPLHLHGFNFPSNFNLVDPMERNTAGVP b)TKAVVLTFNTSVEICLVMQ-GTSIVAAESHPLHLHGFNFPSNFNLVDPMERNTAGVP G-G: 6 H-H: 8
Non-conserved Conserved Sequence profiles ADDGSLAFVPSEF--SISPGEKIVFKNNAGFPHNIVFDEDSIPSGVDASKISMSEEDLLN TVNGAI--PGPLIAERLKEGQNVRVTNTLDEDTSIHWHGLLVPFGMDGVPGVSFPG---I -TSMAPAFGVQEFYRTVKQGDEVTVTIT-----NIDQIED-VSHGFVVVNHGVSME---I IE--KMKYLTPEVFYTIKAGETVYWVNGEVMPHNVAFKKGIV--GEDAFRGEMMTKD--- -TSVAPSFSQPSF-LTVKEGDEVTVIVTNLDE------IDDLTHGFTMGNHGVAME---V ASAETMVFEPDFLVLEIGPGDRVRFVPTHK-SHNAATIDGMVPEGVEGFKSRINDE---- TVNGQ--FPGPRLAGVAREGDQVLVKVVNHVAENITIHWHGVQLGTGWADGPAYVTQCPI TVNGQ--FPGPRLAGVAREGDQVLVKVVNHVAENITIHWHGVQLGTGWADGPAYVTQCPI TKAVVLTFNTSVEICLVMQGTSIV----AAESHPLHLHGFNFPSNFNLVDPMERNTAGVP Matching any thing but G => large negative score Any thing can match
Sequence profiles Align (BLAST) sequence against large sequence database (Swiss-Prot) Select significant alignments and make profile (weight matrix) using techniques for sequence weighting and pseudo counts Use weight matrix to align against sequence database to find new significant hits Repeat 2 and 3 (normally 3 times!)
Example. Sequence profiles • Alignment of protein sequences 1PLC._ and 1GYC.A • E-value > 1000 • Profile alignment • Align 1PLC._ against Swiss-prot • Make position specific weight matrix from alignment • Use this matrix to align 1PLC._ against 1GYC.A • E-value < 10-22. Rmsd=3.3
Sequence profiles Score = 97.1 bits (241), Expect = 9e-22 Identities = 13/107 (12%), Positives = 27/107 (25%), Gaps = 17/107 (15%) 1PLC._: 3 ADDGSLAFVPSEFSISPGEKI------VFKNNAGFPHNIVFDEDSIPSGVDASKIS 56 F + G++ N+ + +G + + 1GYC.A: 26 ------VFPSPLITGKKGDRFQLNVVDTLTNHTMLKSTSIHWHGFFQAGTNWADGP 79 1PLC._: 57 MSEEDLLNAKGETFEVAL---SNKGEYSFYCSP--HQGAGMVGKVTV 98 A G +F G + ++ G+ G V 1GYC.A: 80 AFVNQCPIASGHSFLYDFHVPDQAGTFWYHSHLSTQYCDGLRGPFVV 126 Rmsd=3.3 Å Structure red Template blue
Profile-profile alignment Query Template Compare amino acid preference for the two proteins and pair similar positions (HHpred)
Including structure • Sequence with in a protein superfamily share remote sequence homology • , but they share high structural homology • Structure is known for template • Predict structural properties for query • Secondary structure • Surface exposure • Position specific gap penalties derived from secondary structure and surface exposure
Structure biased alignment (3D-PSSM) http://www.sbg.bio.ic.ac.uk/~3dpssm/
Threading Alignment score from structural fitness (pair potential) How well does K fit environment at P6? If P8 is acidic then fine, if P8 is basic then poor Deletions 7 4 6 2 5 8 10 9 3 1 .. A T N L Y K E T L .. Insertion
Threading • Threading does not work! • The average protein does not exist • Threading can be used in combination with sequence profiles, local structural features to improve alignment
CASP. Which are the best methods • Critical Assessment of Structure Predictions • Every second year • Sequences from about-to-be-solved-structures are given to groups who submit their predictions before the structure is published • Modelers make prediction • Meeting in December where correct answers are revealed
The top 4 homology modeling groups in CASP6 • All winners use consensus predictions • The wisdom of the crowd • Same approach as in CASP5! • Nothing has happened in 2 years!
The wisdom of the crowd! • Why the many are smarter than the few • A general method useful to improve prediction accuracy • No single method or expert will always be the best
The wisdom of the crowd! • The highest scoring hit will often be wrong • Not one single prediction method is consistently best • Many prediction methods will have the correct fold among the top 10-20 hits • If many different prediction methods all have some fold among the top hits, this fold is probably correct
3D-Jury (Best group) • Inspired by Ab initio modeling methods • Average of frequently obtained low energy structures is often closer to the native structure than the lowest energy structure • Find most abundant high scoring model in a list of prediction from several predictors • Use output from a set of servers • Superimpose all pairs of structures • Similarity score Sij = # of Ca pairs within 3.5Å (if #>40;else Sij=0) • 3D-Jury score = SijSij/(N+1) • Similar methods developed by A Elofsson (Pcons) and D Fischer (3D shotgun)
How to do it? Where is the crowd • Meta prediction server • Web interface to a list of public protein structure prediction servers • Submit query sequence to all selected servers in one go • http://bioinfo.pl/meta/
From fold to structure • Flying to the moon has not made man conquer space • Finding the right fold does not allow you to make accurate protein models • Can allow prediction of protein function • Alignment is still a very hard problem • Most protein interactions are determined by the loops, and they are the least conserved parts of a protein structure
Ab initio protein modeling Modelling of new fold proteins • Only when every thing else fails • Challenge • Close to impossible to model Natures folding potential
Challenge. Folding potential • New folds are in general constructed from a set of subunits, where each subunit is part of a known fold. • The subunits are small compared to the overall fold of the protein. No objective function exists to guide the global packing of the subunits. Objective function sij = 120aa dij = 6Å
A way to solution • Glue structure piece wise from fragments. • Guide process by empirical/statistical potential Fragments with correct local structure Natures potential Empirical potential
Example (Rosetta web server) www.bioinfo.rpi.edu/~bystrc/hmmstr/server.php Rosetta prediction Structure
Take home message • Identifying the correct fold is only a small step towards successful homology modeling • Do not trust % ID or alignment score to identify the fold. Use p-values • Use sequence profiles and local protein structure to align sequences • Do not trust one single prediction method, use consensus methods (3D Jury) • Only if every things fail, use ab initio methods
Examples • Iterative Blast • http://www.ncbi.nlm.nih.gov/blast • Sequence
Examples • HHpred