Download
1 / 24
250 likes | 804 Views
Epilepsia y herencia. Pediatría Dr. Sergio Graham. epilepsia. Sócrates, Empédocles , Platón, Moliere, Julio César, Dostoievski, Van Gong, Lord Byron, Napoleón, Carlos V, Alfred Nobel, Lenin y Guadalupe Victoria; tenían algo en común: padecían epilepsia en alguna de sus formas. epilepsia.

E N D
Epilepsia y herencia Pediatría Dr. Sergio Graham
epilepsia • Sócrates, Empédocles, Platón, Moliere, Julio César, Dostoievski, Van Gong, Lord Byron, Napoleón, Carlos V, Alfred Nobel, Lenin y Guadalupe Victoria; tenían algo en común: padecían epilepsia en alguna de sus formas
epilepsia • Proviene del latín: • Ser sobrecogido bruscamente o tomado desde arriba • OMS: “afección crónica de etiología diversa, caracterizada por crisis recurrentes debido a una descarga excesiva de neuronas cerebrales, con pérdida o no de la conciencia, asociadas a diversas manifestaciones clínicas y paraclínicas, que puede ocurrir a cualquier edad”
epilepsia • Uno de los trastornos neurológicos más frecuentes en el mundo, superado por la enfermedad vascular cerebral • 1% población mundial. Aprox 50 millones • En México 1 millón en 2007 • El INNyN prevalencia de epilepsia de 18 casos por cada 1 000 habitantes, la SS 12 casos por cada 1 000 habitantes en el año 2001 • Mortalidad del 4%
epilepsia • El 77% se inicia en la infancia, enfermedad de inicio temprano • El 33% después de los 12 años • Etiologías de epilepsia temprana: traumatismo neonatal, anoxia y sufrimiento fetal • Cuando un consanguíneo tiene diagnóstico de epilepsia tiene 1.5 a 5 veces más predisposición que la población en general
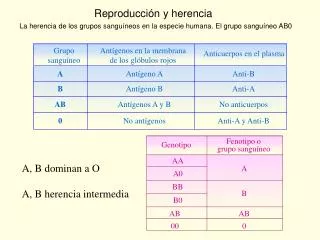
Epilepsia • La herencia de la epilepsia no sigue un patrón mendeliano clásico, y el riesgo de adquirirla sigue un patrón multifactorial • En gemelos monocigóticos la concordancia no es del 100% • El riesgo de transmitirla es mayor cuando es la madre quien la padece (Impronta genómica o mitocondrial) • Heterogeneidad clínica • Pleomorfismo
Genética de las Epilepsias • Epilepsias heredadas de caráterautosómico dominante: • Epilepsia del lóbulo temporal familiar (cromosoma 10) • Epilepsia mioclónica juvenil (cromosomas 6 y 15) • Epilepsia de ausencia en la infancia (croms 1, 3, 5, 6 y 8) • Genes codificadores de canales iónicos neuronales del sodio, potasio, cloro y calcio. • Genes codificadores de los receptores para acetilcolinay GABA. • Otros genes con funciones aún no claramente definidas: EFHC1 y LGI1
SCN1B • Subunidad B-1 de los canales neuronalesde sodio disparados por voltaje • Fase inicial de la despolarización de la membrana sináptica potenciales de acción • Cromosoma 19q13 • GEFS+: Epilepsia Generalizada con Convulsiones Febriles plus • Convulsiones febriles simples o complejas típicas • > 6 años • Convulsiones afebriles • Tónico-clónico generalizadas • Autosómico dominante • Penetranciade hasta 60% • Raro: ausencias, mioclonías o crisis parciales, puede presentarse en forma muy severa como la Epilepsia Mioclónica-Astática o la Epilepsia Mioclónica Severa de la Infancia (EMSI)
SCN1A • Subunidad α-1 de los canales neuronalesde sodio disparados por voltaje • GEFS+2 • Cromosoma 2q21-q33 • Fenotipo heterogeneo
KCNQ2 y KCNQ3 • Miembros 2 y 3 de la subfamilia KQT de los canales de potasio dependientes de voltaje • Cromosoma 20q13.3 • Convulsiones Neonatales Familiares Benignas • Ambos sexos por igual • Crisis el segundo o tercer día de vida, para desaparecer alrededor de la sexta semana • Generalizadas y con componentes tónico-clónicos • 10% síndromes epilépticos clásicos en la edad adulta • Patrón de herencia autosómicodominante • Convulsiones Infantiles Familiares Benignas • Entre los cuatro y ocho meses de edad • Convulsiones en salvas • Mayor preponderancia de manifestaciones focales
CLCN2 • Canales de Cloro disparados por voltaje tipo 2 • Epilepsia idiopática generalizada • Cromosoma 3q26 • Ausencias Típicas de la Niñez, las Ausencias Juveniles, la Epilepsia Mioclónica Juvenil y la Epilepsia Generalizada Tónico-Clónica con crisis al despertar • Mantiene las concentraciones basales de cloro intracelular necesarias para que pueda ocurrir la actividad inhibitoria del neurotransmisor GABA
Cromosoma 6 • Contiene aprox 1900 genes • 170 millones de pares de bases • Enfermedades asociadas a mutaciones: • Ataxia espinocerebelosa • Diabetes • Hperplasia adrenal congénita por deficiencia de 21-hidroxilasa • Epilepsia • EMJ 1 • Hemocromatosis • Síndrome de Zellweger
Epilepsia mioclónica juvenil • Síndrome epiléptico generalizado idiopático • Epilepsia mioclónica más frecuente: 10 % de todas las epilepsias • Antecedentes familiares de epilepsia • Locus en el brazo corto del cromosoma 6(6p21.3)23 • La EMJ «clásica»: mioclonía al despertar, crisis convulsivas de gran mal y EEG con polipunta-ondas de 4 a 6 ciclos por segundo que se presentan en la adolescencia
Epilepsia mioclónica juvenil • Brazo largo del cromosoma 15 (15q14) • Cromosoma 18 • Las manifestaciones clínicas se inician entre los 12 a 18 años • Afectan ambos sexos por igual • Crisis mioclónicas, tónico-clónicas generalizadas y ausencias típicas
Epilepsia mioclónica juvenil • Crisis mioclónicas: sacudidas musculares breves, aisladas o agrupadas que afectan principalmente miembros superiores de forma bilateral • No se acompañan de pérdida de la conciencia. • Generalmente se manifiestan por las mañanas, al despertar o unos minutos más tarde
Epilepsia mioclónica juvenil • 42% mioclonías asimétricas o unilaterales • Crisis tónico-clónicas generalizadas 90% al despertar • El examen neurológico y el intelecto son normales • Factores precipitantes: privación de sueño, el estrés y el consumo de alcohol y fotoestimulación
Epilepsia mioclónica juvenil • El EEG de vigilia puede ser normal o presentar descargas generalizadas de punta-onda y polipunta-onda • Pueden observarse paroxismos con predominio focal en uno y otro hemisferio que pueden conducir a errores diagnósticos de epilepsia parcial • El EEG de sueño muestra en todos los pacientes las anomalías paroxísticas • Estudios neuropsicológicos sugieren disfunciones en el lóbulo frontal
Epilepsia mioclónica juvenil • En espectroscopia por resonancia magnética se observan concentraciones reducidas de N-acetilaspartato en la región prefrontal • Tratamiento de elección: ácido valproico • Lamotrigina empleada en monoterapia o asociada al ácido valproico • Benzodiazepinas (clonazepam o clobazam) son alternativa terapéutica
Epilepsia mioclónica juvenil • Tálamo y el núcleo reticular: marcapasos para las descargas anormales • Regulación circadiana en el número de las descargas: niveles intermedios de vigilancia, durante el sueño lento, en etapas de transición del sueño • Antagonistas GABA disminuyen las descargas de espigas
ATAXIAS CEREBELOSAS AUTOSOMICO DOMINANTES • Enfermedades con diversos fenotipos • Ataxia • Disartria • Dismetría • Temblor intencional acompañado de diversos grados • Ganglios basales, troncoencéfalo, medula espinal, nervio óptico, retina y nervio periférico • Clasificación fenotípica que no corresponde con los hallazgos moleculares
ATAXIAS CEREBELOSAS AUTOSOMICO DOMINANTES • Herencia autosómica dominante • Alteración molecular: • SCA 1 expansión de un trinucleótido CAG mas de 39 veces en el cromosoma 6. La edad de inicio se correlaciona con el nº de repeticiones. Transmisión paterna fenómeno de anticipación • SCA 3 (MJ) expansión de un trinucleótido CAG mas de 64 veces en el cromosoma 14 • SCA 6 mutación en el mismo gen que la migraña hemipléjica familiar y la ataxia episódica tipo 2: CACNL 1A4 situado en el cromosoma 1