Download
1 / 125
1.25k likes | 1.26k Views
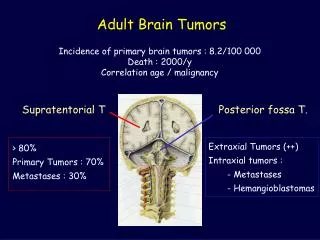
Solid Tumors Dr. H. Murat MUTUŞ. Wilms’ Tumor. Also called as Nephroblastoma One of the most common solid tumors of pediatric age group Seen in frequency of 8/10 6 under the age of 15 years Frequency varies according to geographical areas Age, Sex and Bilaterality:

E N D
Solid Tumors Dr. H. Murat MUTUŞ
Wilms’ Tumor • Also called as Nephroblastoma • One of the most common solid tumors of pediatric age group • Seen in frequency of 8/106 under the age of 15 years • Frequency varies according to geographical areas • Age, Sex and Bilaterality: • Male/Female ratio=1/1,1 • Mean age of admittance is around 3,5-4 years • Bilateral Wilms tumor is seen in 7% of cases
Associated anomalies accompanying Wilms’ Tumor • Aniridia, • Hemihypertrophy • Undescended testis, • Hypospadias • Male pseudohermaphroditism • Renal hypoplasia • Renal ectopia • Duplication anomalies • Horseshoe kidney
Syndromes accompanying Wilms’ Tumor • WAGR syndrome • Wilms’ Tumor, aniridi,a genitourinary malformations, mental retardation • Wiedemann- Beckwith syndrome (WBS) • Macroglossia, organomegaly, hemihypertrophy, abdominal wall defects, hypoglycemia, earbud deformities • Drash syndrome • Pseudohermaphrotidism, degenerative kidney diseases • Klippel-Trenaunay syndrome • Perlmann syndrome • Specific cytogenetic anomalies
Wiedemann- Beckwith syndrome (WBS) • Macroglossia • Organomegaly • Hemihypertrophy • Abdominal wall defects • Hypoglycemia • Earbud deformities
Wilms’ Tumor • Aniridia • 1000 times higher risk for development of Wilms’ Tumor • All children with aniridia should be physically examined and evaluated by abdominal ultrasound every 3 months in first 2 years, snd and every 6 months until 4 years of age • Hemihypertrophy: • 1000 times higher risk when compared with normal population • Such cases should be followed same as in aniridia
Wilms’ Tumor • Genitourinary anomalies • While incidence of undescended testicle and hypospadias are 0,7% and 1,0% in normal population, in Wilms’ tumor cases this ratio is 5,2% • Incidence of bilateral tumors are higher in this group of patients with genitourinary anomalies (25%) • Familial Wilms’ tumor • Family history is 1.5% in Wilms tumor • Bilaterality is higher in familial cases (16%)
Table. Rate of congenital anomalies and syndromes seen in Wilms’ tumor patients. NWTS group results between the years 1980 and 1999.
Molecular Genetics: • Wilms’ tumor gene • WT-1 gene; a tumor supressor gene located in p13 arm of chromosome 11 (11p13). • This gene is deleted in all children with WAGR sendrome. In sporadic Wilms’ tumor patients, deletion is seen in 5-10 % of cases. • WT-2 gene (11p15) is also a supressor gene. • Other genetic anomalies are seen. • Eg: p53 supressor gene plays a role in aggressive form of Wilms’ tumor.
Clinical findings / Symptoms • Abdominal mass • Generally first sign • Hypertension • Hematuria
Clinical findings / Symptoms • A large, regular surfaced, smooth mass may be palpated • The lesion generally is not painful, and is relatively immobile • Generally does not cross the midline • Abdominal pain, fever, and anemia may occur • intraperitoneal or subcapsular bleeding is a result of tumor necrosis. Therefore, when this occurs, it is difficult to differentiate from Neuroblastoma • İntravenous involvement • cardiac murmur, hepatosplenomegaly, ascites, varikocele and gonadal metastasis • Generally metastasizes to lungs
DIAGNOSIS / Imaging • Routinely; • Chest X-Ray • Abdominal plain graphy, followed by • USG and CT and/or MRI • Nature of the mass, and the organ of origin, • Contralateral renal parenchymal evaluation, • Presence of Bilateral disease, • Patency of VCI ve renal vein • Presence of distant metastasis.
Pathology • Composed of blastemal, stromal or epithelial elements. • Any cell type may be dominant, or there may be mixed tissue pattern • Histopathologically; “favorable or unfavorable histology” / The most important prognostic factor
Pathology • Nephroblastomatosis (Nephrogenic rest) • accepted as a premalignant lesion • Histologically it is a persistant metanephric tissue • Has a clear relationship with Wilms’ tumor • They are found in 1% of postmortem autopsies of normal population, but seen as frequent as 25-40 % of Wilms’ tumor patients • They are the most important findings for increased risk of future contralateral Wilms’ tumor occurance
Wilms T-NWTS-5 Staging System • Stage I: Tumor limited to kidney and totally resectable – Renal capsule is intact • Stage II: Tumor extending beyond kidney, but still totally resectable • Stage III: Residual nonhematogenous tumor present, but limited to abdomen • Stage IV: There is hematogenous or lymph node metastasis extending beyond abdominopelvic area • Stage V: There is bilateral kidney involvement during diagnosis
Stage 1 Tumor limited to kidney and totally resectable – Renal capsule is intact
Stage 2 Tumor extending beyond kidney, but still totally resectable
Stage 3 Residual nonhematogenous tumor present, but limited to abdomen
Stage 4 There is hematogenous or lymph node metastasis extending beyond abdominopelvic area
Stage 5 There is bilateral kidney involvement during diagnosis
Surgical Treatment • 2 basicresponsibility of surgeonexists: • Complete resection of the WHOLE viabletumortissue, • Supplycorrectinformationrequiredforstaging Inmostcasesunilateralradicalnephroureterectomy is performed The presence of venousinvolvementorcontralateraldiseaseshould be evaluated, andifdiagnosed, specificsurgicalproceduresshould be done accordingly Continuedclinicalstudiesnamed “NationalWilms’ Tumor Study-1,2,3,4,5” developtreatmentmodalities of radiationandchemotherapy . Theseprotocolsareused as adjuvantstosurgicaltreatment.
CLEAR CELL SARCOMA • Known as bone metastasizing renal tumor in children. They resemble to WT with UH. 25% of cases metastasize to bone. • Important histological features; fibrovascular septae formation in tumor with vascular arcus formation and division of tumor cells into chords or columns • Multicentric or bilateral clear cell sarcoma has not been reported • In treatment all patients undergo nephrectomy, radiotheraphy, and chemotheraphy as a standart approach. If bone metastasis present, long term survival is very low (16%)
RHABDOID TUMORS • Rare but very fatal • They are thought to origin from neural crest cells of renal medulla • Consist of %2 of renal tumors • Hypercalcemia may be seen • Differential diagnosis from WT is possible by CT has been reported • All cases are unilateral • May metastasize to anywhere, but most frequently to lungs • Surgery, chemotherapy and radiotherapy are used for treatment, but results are not satisfactory
RENAL ADENOCARCINOMA • They are very rare tumors in children. Most frequently seen in late childhood, or in adolescents. • In adult forms, the triad of abdominal pain, hematuria, and flank mass is classical; but in children and adolescents, this is not so common. • Metastasize to lungs, liver, and bone. Prognosis depends on clinicopathological stage. • Recommended treatment is; radical nephrectomy+regional lymphadenectomy. Expected survival rate is around 50%. Chemotherapy and radiotherapy may also be added to treatment.
CONGENİTAL MESOBLASTİC NEPHROMA • Most common renal tumor in first 3 months of life • After 4. month, Wilms’ tumor is most common • 5% of all pediatric renal tumors • More than %90 are seen in 1st year of life • Twice more common in male children • Most common age of admittance is between 3-4 months of age
CONGENİTAL MESOBLASTİC NEPHROMA • The most common presenting sign is a palpable mass in the abdomen • Hematuria may rarely be seen • May be diagnosed prenatally • Polihydramnios may be present • Preterm delivery, low birth weight, and hydrops faetalis may occur
CONGENITAL MESOBLASTIC NEPHROMA • No Bilateral lesion reported • Treatment is surgical removal • %95 cure with surgery only, • %5 recurrence possible • USG follow up after surgical removal is necessary • Although this tumor has a relatively benign character, it may rarely act agresively
CONGENITAL MESOBLASTIC NEPHROMA Longitudinal section of kidney showing large blood clot occupying upper and middle poles with thinned cortex
CONGENİTAL MESOBLASTİC NEPHROMA Low-power photomicrograph showing cystic area, large fibrous strand, and innumerable small, round cells in renal cortex typical of mesoblastic nephroma