Download

1 / 40
410 likes | 725 Views
Palliative care in motor neuron disease. Gary Hsin, M.D. November 18 th , 2005. Using ALS as a model for discussion. Amyotrophic Lateral Sclerosis. Progressive, degenerative neurologic disease of unknown etiology Involves both upper and lower motor neurons

E N D
Palliative care in motor neuron disease Gary Hsin, M.D. November 18th, 2005
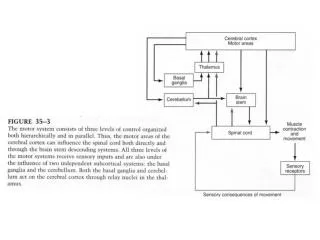
Amyotrophic Lateral Sclerosis • Progressive, degenerative neurologic disease of unknown etiology • Involves both upper and lower motor neurons • LMN: weakness, atrophy, fasiculation due to denervation of muscles – amyotrophic • UMN: hyperreflexia, spasticity due to lateral corticospinal tract degeneration – lateral sclerosis
ALS continued • Muscle atrophy and spasticity in limb and bulbar muscles result in weakness and loss of ambulation, oropharyngeal dysfunction, weight loss, and ultimately respiratory failure. • The mainstay of treatment for ALS patients is still palliative and symptomatic management. • It is important to provide anticipatory guidance, and continue the discussion of goals of care.
Pathology & Etiology • Motor neuron degeneration and death • Superoxide dismutase type 1 mediated toxicity • Excitotoxicity via glutamate mediated signaling • Cytoskeletal derangements, mitochondrial dysfunction, viral infections, apoptosis
Epidemiology & Prognosis • 5000 new case in the U.S. per year • 90% cases sporadic, 10% familial • Incidence 1.47~2.7 per 100,000 per year • Prevalence 2.7~7.4 per 100,000 per year • 70% die within 3~5 years of diagnosis • 10~20% survive for more than 10 years • Improved survival associated with younger age of onset, male gender and limb rather than bulbar symptoms
Riluzole • Riluzole reduces the presynaptic release of glutamate, but the precise mechanism of action in ALS is unclear. It is postulated that it may be neuroprotective by reducing excitotoxicity that is due to excess glutamate, and it may also act as a glutamate receptor antagonist.
Riluzole continued • Clinical trials so far do show that Riluzole improved survival, especially in younger populations, but demonstrated no beneficial effect on bulbar function or muscle strength • The pts who are most likely to benefit are those who have symptoms for less than 5 years, vital capacity >60%, and no tracheostomy. 50mg PO BID.
Signs & Symptoms • Limb spasticity • Hyperreflexia • Brisk jaw reflex • Babinski's sign • Pseudobulbar palsy with emotional lability • Focal or multifocal limb weakness & atrophy • Cramps & fasciculations • Dysarthria, dysphagia and/or choking • Tongue atrophy with fasciculations • Claw hand • Fatigue • Dyspnea
Secretion management • In ALS there is overall decreased saliva production, sialorrhea is more a poor handling of saliva. • The management is similar for pts with cerebral palsy, MR, oropharyngeal carcinoma, Down syndrome. • Decrease saliva production, or divert and remove saliva
Secretion management • Glycopyrrolate (Robinul) 1~2mg BID or TID • Benztropine (Cogentin) 0.5~2.0mg QD or BID or Atropine 0.4mg Q4~6 hours • Amitriptyline (Elavil) 10~150mg QHS • Transdermal scopolamine one or two patches every three days • Trihexyphenidyl (Artane) 1 to 2 mg per day slowly increasing to total of 5 to 15 mg daily in three or four divided doses • May also try Clonidine, Levsin
Secretion management • Botulinum toxin injection into the salivary glands appears to be safe and useful in preliminary studies • External beam radiation [3-30 Gy; 3-10 fractions] • Surgical interventions have been tried w/o demonstrated efficacy
Secretion management • Thick secretions/mucus • Increase fluid intake • Humidified air • Propranolol or Metoprolol
Secretion management • Non-pharmacologic management • Suction machine (not usually helpful for thick mucus, but helpful with sialorrhea) • Mechanical insufflation-exsufflation (In-Exsufflator cough machine) • Manually assisted coughing techniques
Muscle spasm & weakness • Early PT/OT involvement • Variety of assist devices may provide, mobility, support, comfort and increase functionality, i.e. braces, wheelchair, special air beds, head rests, etc.
Muscle spasm & weakness • Cramps • Quinine sulfate 200 mg twice a day • Tizanidine 2 to 4 mg by mouth twice daily up to a total dose of 24 mg daily • Carbamazepine 200 mg twice daily • Phenytoin 100 mg once to three times a day • For mild symptoms: • Magnesium 5 mmol once to three times a day • Vitamin E 400 IE twice a day
Muscle spasm & weakness • Spasticity • Baclofen 5 to 10 mg twice daily to three times daily • Tizanidine 2 to 4 mg by mouth twice daily up to a total dose of 24 mg daily • Memantine starting at 5 mg daily, increasing by 5 mg a week to a maximum of 20 mg twice a day • Tetrazepam 50 mg at bedtime, increasing by 25 mg a day to a maximum dose of 150 mg taken two to three times a day
Pseudobulbar affect • Also known as: pseudobulbar palsy, emotional incontinence, pathologic crying/laughing • The emotional lability is NOT a mood disorder, but is an uncontrolled outburst and is a very troubling symptom for patients. • It is an abnormal affective display that can be seen in about 50% of ALS patients.
Pseudobulbar affect • Amitriptyline 10~150mg QHS • Fluvoxamine (Luvox) 100~200mg QD • Alternatively may try Lithium or L-Dopa
Constipation • Common symptom due to immobility and side effects from opioid, anti-cholinergics, muscle relaxants
Pain • Often results from muscle contracture, joint stiffness, and immobility leading to pressure ulcers at later stage of disease
Sleep problems • Often associated with other symptoms • Identify and treat underlying causes • Use sedative cautiously
Dysarthria • Speech therapy often helpful early • Computer technology offer many options to assist with patient communication
Dysphagia and nutrition • Decreased caloric and fluid intake may lead to worsening of symptoms, such as weakness, muscle atrophy, fatigue • Initially management includes the modification of food and liquid consistency • Discussion of possible PEG placement
Dysphagia and nutrition • For optimal safety and efficacy, PEG placement should be performed before the forced vital capacity falls to below 50% of predicted and not in the preterminal phase. • The indications should be to evaluate for symptoms of hunger, choking, sign of inadequate oral intake, and diminished quality of life, rather than a swallow evaluation.
Dysphagia and nutrition • Complications for PEG include: • Transient laryngeal spasm 7.2% • Localized infection 6.6 • Gastric hemorrhage 1~4% • Death 1.9% • Does NOT prevent aspiration pneumonia • Studies suggest that with PEG survival is prolonged by 1~4 months
Respiratory care • It is important to initiate discussion regarding the patient’s goals and how those goal can be best achieved with respect to respiratory care including non-invasive and invasive mechanical ventilation. • Respect the right of patients to refuse or withdraw treatment.
Respiratory care • A decrease of VC to <50% is often associated with respiratory symptoms. • A VC of less than 1L or 25~30% of predicted is associated with significant risk of respiratory failure and sudden death. • There are no current guidelines on how frequently to test VC.
Respiratory care • Dyspnea • Body position such as elevation is often helpful • Chest physiotherapy in the early phase • Opioid can be used to treat dyspnea and benzodiazepines for associated anxiety • Inhaled opioids have not consistently demonstrated benefit; inhaled lidocaine appears to be more efficacious • Oxygen can be administered for hypoxia
Respiratory care • Months to years before terminal respiratory failure, symptoms of chronic nocturnal hypoventilation frequently presents and impair the patient’s quality of life. • The symptoms include: daytime fatigue, concentration difficulty, headache, disturbed sleep, nightmares, nervousness, depression, anxiety, tachypnea, tachycardia, diaphoresis, dyspnea, phonation difficulties, reduced appetite, weight loss, gastritis, cyanosis, edema, recurrent URI, dizziness, syncope, visual disturbance, diffuse pain
Respiratory care • Non-invasive intermittent ventilation administered at night for 4 hours is an efficient way of alleviating nocturnal symptoms. • It should be made clear that this effort is for the purpose of symptom management and improving the quality of life rather than prolongation.
Respiratory care • It is important to explore other means of ventilatory support should it be desired. The goals of such support and when to discontinue such therapies when the goals are no longer being met. Patients need to be reassured that all necessary care will be provided to ensure their comfort. • Non-invasive: Negative pressure body ventilators and oscillators, Intermittent abdominal pressure ventilator, Intermittent positive pressure ventilators (mouth/nasal) • Tracheostomy & ventilation
Respiratory care • Patients show higher satisfaction with NIV than IV. • Studies show that ventilator dependent patients are not more depressed than patients who are not vent dependent, and can lead meaningful lives. • But there is greater financial, social and emotional burden.
Other symptoms • Urinary frequency/urgency • In the absence of UTI, often due to spasticity that responds well to Oxybutinin • Peripheral edema • Often dependent: elevation, massage, compression hose (r/o DVT) • Laryngospasm • Sudden reflex closure of vocal cords due to variety of stimuli, usually resolves spontaneously • H1 and H2 blocking agents may be helpful
Psychosocial and spiritual care • The role of continued support for patient and family cannot be underestimated or overstated. • Bereavement and grief counseling from diagnosis
Additional resources • ALS Association (www.alsa.org) • Muscular Dystrophy Association (www.mdausa.org) • Links to resources for patients and families • Books: • Learning to Fall: The Blessings of an Imperfect Life, Philip Simmons • Tuesdays with Morrie, Mitch Albom
Special considerations in pediatricsusing SMA (spinal muscular atrophy) as a model