Download

1 / 29
360 likes | 676 Views
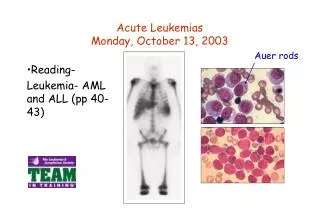
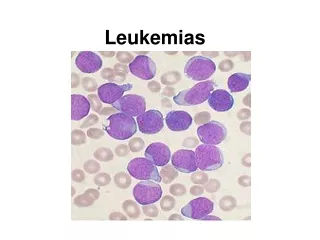
Acute Leukemias. Oyekunle AA MBBS FMCPath. Senior Lecturer & Consultant Haematologist. Acute Lymphoblastic Leukemia. Definition. ALL is a clonal lymphoproliferative disorder of immature lymphocytes aka lymphoblasts >25% in the blood/marrow

E N D
Acute Leukemias Oyekunle AAMBBS FMCPath. Senior Lecturer & Consultant Haematologist
Definition • ALL is a clonal lymphoproliferative disorder of immature lymphocytes aka lymphoblasts • >25% in the blood/marrow • It is characterized by impaired cellular maturation at the early phase of haematopoietic differentiation
Epidemiology • Gender • The incidence of ALL is slightly higher in males than females • Age • 60% of cases are seen in children, with a peak incidence in the first 5 years of life and • A subsequent drop in incidence until age 60 • At 60 years, a second peak emerges • Race and ethnicity • The incidence of acute leukemia is slightly higher in Europeans.
Aetiologic factors There is wide diversity in the behavior of the various subsets of acute leukemias. Thus, it is unlikely that there is one common etiology for these disorders. There are, however, some accepted risk factors for leukemogenesis: • Chemical exposure • Prolonged exposure to benzene & petroleum products. The interval between exposure and onset of leukemia is long (10-30 years). Chromosomal damage is common. • Pesticide exposure. • Exposure to hair dyes, smoking & nonionic radiation may also increase the risk of leukemia.
Prior chemotherapy or irradiation • Use of alkylating agents, such as cyclophosphamide (Cytoxan, Neosar) and melphalan (Alkeran). Cytogenetic abnormalities, particularly monosomy 5, 7, 11, and 17, are common. • Concurrent radiation exposure slightly increases the risk of leukemogenesis posed by alkylating agents. • Topoisomerase II inhibitors (etoposide, teniposide [Vumon], doxorubicin and its derivatives, and mitoxantrone [Novantrone]), have also been linked with leukemogenesis. Cytogenetic abnormalities involving chromosome 11q23 or 21q22 are common. • Genetic disorders • In identical twins younger than age 10, if one child develops leukemia (usually ALL), there is a 20% chance that the other twin will develop leukemia within a year; subsequently,the risk falls off rapidly and joins that of nonidentical siblings, which is 3-5 times that of the general population.
Classification of ALL • May be pre-B or pre-T in origin • Lymphoblasts: Blood or bone marrow ≥25% • cf lymphoblastic lymphoma, with </>25% marrow blasts + nodal tumour • The classical FAB subtypes (L1-L3) are no longer recognized by the new WHO classification • ALL-L3 is now classified as Burkitt’s lymphoma (BL) because it expresses smIg, does not express TdT & has the same chromosomal abnormalities as BL. • Classification is now based on morphologic, immunologic and cytogenetic characteristics (see Table)
Precursor B-cell ALL • The majority of ALL • There is expression of CD19 and CD 10 (common ALL antigen, cALLa) • Chromosome abnormalities are often seen • Mature B-cell (Burkitt-cell) ALL • The least common of ALL, formerly ALL-L3 • Has the worst prognosis • Associated with c-myc on ch8 & • Ig heavy-chain gene on ch14 t(8;14), in 80% of cases or • Light-chain genes on ch2 t(2;8) or 22 t(8;22), in the other 20% • Precursor T-cell ALL • Associated with translocation or deletion of the ch 9 (t or del 9p) • Prognosis is generally poor • T-cell ALL • Frequently associated with translocations of TCR genes on ch 11 or 7 • Prognosis is very poor with conventional chemotherapy • May be seen in some individuals infected with HTLV-1
Table 1: Morphologic, immunologic and cytogenetic classification of ALL
Prognostic factors in ALL • High-risk ALL • Age <1 or >10 years • Male sex • Leucocyte count >50 x 109/l • Poor performance status • Failure of primary induction • Presence of t(9;22), t(1;19) • B-,T- or mixed lineage phenotype • CNS disease at diagnosis (ie, presence of at least 5 leucocytes/uL of CSF plus blasts in a cytocentrifuged smear and/or the presence of cranial nerve palsies) • Massive lymphadenopathy and/or hepatosplenomagaly • MLL rearrangement • Good-risk ALL • Complete remission (CR) within 14 days of induction therapy • Pre-B ALL • Children ages 1-9 • Leucocyte count < 10 x 109/l at diagnosis • Presence of hyperdiploidy (> 50 chromosomes) • Absence of CNS disease or testicular disease at presentation • Absence t(9;22)-Ph+ bcr/abl p190 or t(1;9) translocations • Absence of MLL rearrangement • Standard-risk ALL • T-cell ALL and pre-B ALL that do not meet the criteria for low- or high-risk
Clinical Features • Features of bone marrow failure • Anaemia: easy fatigability • Thrombocytopenia: bleeding • Leukocytosis • But usually functional neutropenia: fever • Hyperleukocytosis when WBC ≥100,000/μl • Increased risk of CNS disease • Tumor lysis syndrome • Leukostasis • Infiltration by blasts • CNS, Skin, GIT, GUS, Resp, CVS etc. • Some are comatose/blind from intracerebral/retinal bleeding • Testicular swelling in 2% (especially in T-ALL); acts as a “sanctuary” site, usually responsible for early relapse • Bone pain • Especially in children; from packing of the marrow
Investigations • FBC: abnormalities raise the possibility of leukemia • BMA: substantiates the diagnosis • Cytochemistry • Immunophenotyping • Fluorescent-activated cell sorter (FACS) using mAb directed at leukemia-specific antigens, and • Cytogenetic analysis of the marrow or peripheral blood blasts at diagnosis • Coagulation profiles: • PT, PTR & INR • aPTT (PTTK) • Serum E&U, Cr & LFT • Used to evaluate metabolic abnormalities • Lumbar puncture in all patients at diagnosis regardless of CNS status • For CSF cytology
Management: Supportive • Counselling & psychosocial support • Prevention of tumor lysis syndrome • Hydration & • Allopurinol &/or Uricase • Transfusion support • Aggressive infection surveillance & treatment • Close watch on renal & hepatic function parameters • Attention to adequate nutrition • Pain control
Management: Definitive • Pre-B and T-lineage ALL are treated similarly, with 3 distinct phases: • Induction remission, • Intensification/Consolidation and • Maintenance. • Vincristine & glucocorticoid (prednisolone or dexamethasone) are key to induction therapy • Because most of our patients have high-risk disease, anthracycline & L-asparaginase are needed in order to achieve durable remission. • But asparaginase is rarely affordable to most patients • In practice, the more readily affordable cyclophosphamide and ara-C are added to the standard vincristine and glucocorticoid (COAP regimen).
Dexa is preferred to Pred because of its greater CNS penetration & lymphoblastocidal action • T-ALL carries a poorer prognosis, but may do well with the inclusion of Cyclophosphamide & Ara-C • The mature B-cell ALL has the worst prognosis • Usually, remission maintenance is not needed in B-ALL because relapse is rarely seen after the 1st yr of induction • CNS prophylaxis with Ara-C, Mtx &/or Hydrocortisone is indicated in all cases of ALL • Older ALL patients (>60 years) fare worse in all ALL subtypes
Treatment Plan B. Intensification Therapy Intensification therapy starts day 14 to day 28 of CR. Two cycles of higher intensity regimens or the same agents used for induction therapy (1-3 above) are given at 1 month interval C. Maintenance Therapy (2-3 years) Girls should have ≥2-2.5yrs of maintenance therapy & boys ≥3 yrs. Adults should be on continuous therapy for 2 years. • 6-mercaptopurine 100 mg/m2 PO every day plus • Mtx 20 mg PO or IV Every Week D. CNS Therapy in Pre-B & T-lineage ALL CNS prophylaxis: IT Mtx 12 mg or Ara-C 25mg days 1 & 5 of therapy weekly until CR, • Starting 2nd week of induction therapy In the presence of CNS leukaemia; • Triple CNS therapy: 12 mg MTX plus 25mg ara-C plus 50 mg hydrocortisone Days 1 & 5 until CNS remission A. Induction Therapy (1 or 2 cycles) OAP • Oncovin 1.5 mg/m2 IV days 1, 8, 15 & 22 • Adriamycin 25 mg/m2 IV days 1, 8, 15 & 22 • Prednisolone 40 mg/m2 (or dexamethasone 6 mg/m2) PO days 1-28 OR OAPL • Oncovin 1.5 mg/m2 IV days 1, 8, 15 & 22 • Adriamycin 25 mg/m2 IV days 1, 8, 15 & 22 • Prednisolone 40 mg/m2 (or dexamethasone 6 mg/m2) PO days 1-28 • L-Asparaginase 5,000 IU/m2 IV days 1-14 OR COAP • Cyclophosphamide 650 mg/m2 IV days 1 & 8 • Oncovin 1.5 mg/m2 IV days 1, 8, 15 & 22 • Ara-C 100 mg/m2 IV continuous infusion days 1-7 • Prednisolone 40 mg/m2 (or dexamethasone 6 mg/m2) PO days 1-28
Treatment Plan: Transplantation • Ph+ & other high-risk ALL • Allogeneic HSCT (allo-HSCT) is advised in 1st complete remission (CR1) • Cure rates of approx 55% are expected • Standard-risk ALL • Usually considered for allo-HSCT in CR2 • Cure rates of up to 50% have been reported • Autologous HSCT (auto-HSCT) • Associated with lower transplant-related mortality (TRM) but higher relapse rates compared to allo-HSCT • Overall survival (OS) is however worse than in AML • An OS of ~28% has recently been reported in patients with refractory disease with allo-SCT
Definition • A malignant clonal disorder of immature cells in the haematopoietic hierarchical system • Arise from multiple hits at the molecular level, the phenotypic expression is found as; • a proliferative lesion • defective differentiation • a resistance to apoptosis
Epidemiology • Commonest leukemia in the neonatal period • 2–3/100,000/yr in UK children • 15–20% of acute leukemias • 15/100,000 in UK adults • 80% of acute leukemias • M : F = 2 : 1
Associations exist with Irradiation Smoking Chemical exposure (benzene, pesticides, herbicides) Inherited conditions Downs syndrome Ataxia telangiectasia Blooms syndrome Fanconi anaemia Neurofibromatosis Schwachmann Syndrome Unknown in most cases Clonal evolution or predisposing diseases Essential thrombocythaemia Polycythaemia vera Myelofibrosis Chronic myeloid leukemia Paroxysmal nocturnal haemoglobinuria Aplastic anaemia HIV/AIDS Multiple myeloma Aetiologic factors
Table 1: FAB classification of AML subtypes using monoclonal antibodies AML = acute myelogenous leukemia; APL = acute promyelocytic leukemia; CD = cluster of differentiation; HLA-DR = human leukocyte antigen D-related; MPO = myeloperoxidase; PAS = periodic acid-Schiff; Tdt = terminal deoxynucleotidyl transferase.Adapted from O’Donnel MR. Acute leukemias. In: Cancer Management: A multidisciplinary approach. pp 769-96
Clinical Features • Features of bone marrow failure • Anaemia: easy fatigability • Thrombocytopenia: bleeding • Coagulopathies; can also occur in AML-M3, M4 or M5 • Leukocytosis • But usually functional neutropenia: fever • Hyperleukocytosis when WBC ≥100,000/μl • Increased risk of CNS disease • Tumor lysis syndrome • Leukostasis • Infiltration by blasts • Skin, GIT, GUS, Resp, CVS, CNS etc. • Massive hepatosplenomegaly usually suggests prior dx • Bone pain • Especially in children; from packing of the marrow
Investigations • FBC: abnormalities raise the possibility of leukemia • BMA: substantiates the diagnosis • Cytochemistry • Immunophenotyping • Fluorescent-activated cell sorter (FACS) using mAb directed at leukemia-specific antigens, and • Cytogenetic analysis of the marrow or peripheral blood blasts at diagnosis • Coagulation profiles: • PT, PTR & INR • aPTT (PTTK) • Serum E&U, Cr & LFT • used to evaluate metabolic abnormalities • Lumbar puncture at diagnosis in all patients with neurologic symptoms regardless of age & pathology
Management: Supportive • Counselling & psychosocial support • Prevention of tumor lysis syndrome • Hydration & • Allopurinol &/or Uricase • Transfusion support • Aggressive infection surveillance & treatment • Close watch on renal & hepatic function parameters • Attention to adequate nutrition • Pain control
Management: Definitive • Definitive treatment of AML is done in 2 distinct phases: • Induction remission and • Consolidation • Ara-C & the anthracyclines are the mainstay of initial therapy for AML • Idarubicin is replacing Daunorubicin as the anthracycline of choice to pair with Ara-C • In practice however, the more readily available COAP regimen are used locally
Treatment Plan Chemotherapy (CT) alone can achieve a disease-free-survival (DFS) of 10-35% at 5 years. A. Induction Therapy (1-2 cycles) AI • Ara-C 200 mg/m2 IV days 1-7 • Idarubicin 12 mg/m2 IV days 1-3 OR COAP • Cyclophosphamide 650 mg/m2 IV days 1 & 8 • Oncovin 1.5 mg/m2 IV days 1 & 8 • Ara-C 100 mg/m2 IV continuous infusion x days 1-10 • (or 50 mg/m2 SC 12hrly x 10 days) • Prednisolone 40 mg/m2 (or dexamethasone 6 mg/m2) PO days 1-10 B. Consolidation Therapy • Ara-C 3 g/m2 q12h IV as 2-3 hr infusion on days 1, 3, 5; repeat q28d × 4 cycles All-trans-Retinoic acid (ATRA) is usually incoporated for remission induction, in the management of AML-M3 (APL).
Table 2: AML induction and consolidation therapy ALSG = Australian Leukemia Study Group; AML = acute myeloblastic leukemia; Ara-C = cytarabine; Daun = daunorubicin; IDA = idarubicin; VP-16 = etoposidea Idarubicin has been substituted for Daun, 45 mg/m2, which had been the prevalent anthracycline used in clinical trials prior to 1993. Mitoxantrone, 10 mg/m2 × 5 days, has also been used as an alternative.b For patients < 60 years of ageAdapted from O’Donnel MR. Acute leukemias. In: Cancer Management: A multidisciplinary approach. pp 769-96
Treatment Plan: Transplantation • Best results for AML with allogeneic SCT (allo-SCT) are in 1st complete remission (CR1), with average cure rates (DFS at 5 years) of 60-65% • However in CR2 or 1st relapse (R1), it drops to 30-35% • Intermediate & high-risk patients with an HLA-matched donor are offered allo-SCT in CR1 • Patients in CR1 lacking a matching donor may be offered CB-SCT or T-depleted haplo-identical SCT • Auto-SCT can be used to delay disease progression when matching donors are unavailable • However, low-risk patients can be transplanted in CR2 • In AML-M3 (APL), cure rates of >70% can be achieved using standard CT alone • HSCT is only advised when CT has failed