Download

1 / 22
230 likes | 607 Views
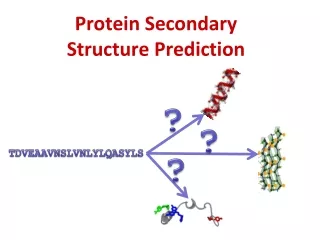
Secondary structure prediction from amino acid sequence. Homology: Paralogs and orthologs . a. duplication. a. b. Paralogs = gene families in same species. speciation. a. b. a. b. orthologs. species 1. species 2. DNA sequence. Automatic translation.

E N D
Homology: Paralogs and orthologs a duplication a b Paralogs = gene families in same species speciation a b a b orthologs species 1 species 2
DNA sequence Automatic translation Physico-chemical properties (e. g., using EMBOSS suite) Amino acid primary sequence Primary db searches FASTA, BLAST 1. Search for sequence homologue(s) and construct an alignment 2. Homologue(s) with known 3D structure? Homology modelling available 3. Motif recognition: Search secondary databases Secondary structure prediction Fold assignment
Chou-Fasman Parameters • Amino acid propensities
Accuracy of prediction • Q3 score Q3 = qa+qb+qcoil X 100% total no. of residues
Recent improvements • The availability of large families of homologous sequences has greatly enhanced secondary structure prediction. • The combination of sequence data in multiple alignments with sophisticated computing techniques such as neural networks has lead to accuracies well in excess of 70 %. • The limit of 70-80% may be a function of secondary structure variation within homologous proteins.
Stereochemical analysis Patterns of residue conservation are indicative of particular secondary structure types. Alpha helices have a periodicity of 3.6. Many alpha helices in proteins are amphipathic, meaning that one face is pointing towards the hydrophobic core and the other towards the solvent. Patterns of hydrophobic residue conservation showing the i, i+3, i+4, i+7 pattern are highly indicative of an alpha helix. XOOXXOOX
Stereochemical analysis The geometry of beta strands means that adjacent residues have their side chains pointing in oppposite directions. Beta strands that are half buried in the protein core will tend to have hydrophobic residues at positions i, i+2, i+4, i+8 etc, and polar residues at positions i+1, i+3, i+5, etc. XOXOXOXOXO
Stereochemical analysis Beta strands that are completely buried (as is often the case in proteins containing both alpha helices and beta strands) usually contain a run of hydrophobic residues. XXXXXXXXXXXX
Helical transmembrane proteins + • Strong hydrophobicity signal from membrane spanning regions, each ~25 residues in length • Predominance of positively charged amino acid residues on cytoplasmic side • Prediction accuracy with multiple alignment = 95%
Helical transmembrane proteins • ~30% of top 100 drugs bind to membrane proteins • Difficult to determine experimentally • But much easier to predict than globular proteins! • TMpred – based on statistical analysis of transmembrane proteins • TMHMM – based on Hidden Markov Model
Protein Structure Classification Class(C) secondary structure content – mainly alpha, mainly beta, alpha/beta, few secondary structures (type) Architecture(A) gross arrangement of sec. structure elements (type and number of SS elements) Topology(T) shape and connectivity of SS (type, number and order of SS elements) Homologous superfamily (H) http://www.cathdb.info/latest/index.html
Homologous domains, share common ancestor Fold families Class Architecture Topology H-level
In CATH, the assignments of structures to fold groups and homologous superfamilies are made by sequence and structure comparisons. Homologous domains, share common ancestor Fold families Class Architecture Topology H-level
Homologous domain family ? Architecture: ‘Barrel’ 9 Topologies : type of SS, number and order
Secondary structure prediction methods • PSI-pred (PSI-BLAST profiles used for prediction; David Jones, Warwick) • JPRED Consensus prediction (includes many of the methods given below; Cuff & Barton, EBI) • DSC King & Sternberg • PREDATORFrischman & Argos (EMBL) • PHD home page Rost & Sander, EMBL, Germany • ZPRED server Zvelebil et al., Ludwig, U.K. • nnPredict Cohen et al., UCSF, USA. • BMERC PSA Server Boston University, USA • SSP (Nearest-neighbor) Solovyev and Salamov, Baylor College, USA. http://speedy.embl-heidelberg.de/gtsp/secstrucpred.html
Consensus prediction method hydrophobic highly conserved b= buried, e = exposed
Consensus prediction method -JPRED hydrophobic amphipathic hydrophobic highly conserved b= buried, e = exposed
Neural network prediction - PHD Multiple alignment of protein family SS profile for window of adjacent residues
Hidden Markov Models-HMMSTR amino acid secondary structure element structural context Markov state • Recurrent local features of protein sequences • Accuracy of 74% • Bystroff et al., 2000