Download

1 / 33
380 likes | 603 Views
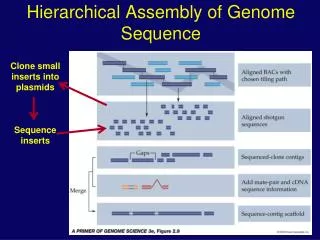
Genome sequence assembly. Assembly concepts and methods (some slides courtesy of Mihai Pop). Building a library. Break DNA into random fragments (8-10x coverage). Actual situation. Building a library. Break DNA into random fragments (8-10x coverage) Sequence the ends of the fragments

E N D
Genome sequence assembly Assembly concepts and methods (some slides courtesy of Mihai Pop)
Building a library • Break DNA into random fragments (8-10x coverage) Actual situation
Building a library • Break DNA into random fragments (8-10x coverage) • Sequence the ends of the fragments • Amplify the fragments in a vector • Sequence 800-1000 (500-700) bases at each end of the fragment
I II R F I II R F II I R F Forward-reverse constraints • The sequenced ends are facing towards each other • The distance between the two fragments is known (within certain experimental error) Insert R F Clone
Building Scaffolds • Break DNA into random fragments (8-10x coverage) • Sequence the ends of the fragments • Assemble the sequenced ends • Build scaffolds
Assembly gaps Physical gaps Sequencing gaps sequencing gap - we know the order and orientation of the contigs and have at least one clone spanning the gap physical gap - no information known about the adjacent contigs, nor about the DNA spanning the gap
Unifying view of assembly Assembly Scaffolding
Typical contig coverage Imagine raindrops on a sidewalk
Lander-Waterman statistics L = read length T = minimum detectable overlap G = genome size N = number of reads c = coverage (NL / G) σ = 1 – T/L E(#islands) = Ne-cσ E(island size) = L((ecσ – 1) / c + 1 – σ) contig = island with 2 or more reads
Example Genome size: 1 Mbp Read Length: 600 Detectable overlap: 40
Experimental data Caveat: numbers based on artificially chopping up the genome of Wolbachia pipientis dMel
Read coverage vs. Clone coverage 4 kbp 1 kbp Read coverage = 8X Clone (insert) coverage = 16 2X coverage in BAC-ends implies 100x coverage by BACs (1 BAC clone = approx. 100kbp)
Assembly paradigms • Overlap-layout-consensus • greedy (TIGR Assembler, phrap, CAP3...) • graph-based (Celera Assembler, Arachne) • Eulerian path (especially useful for short read sequencing)
TIGR Assembler/phrap Greedy • Build a rough map of fragment overlaps • Pick the largest scoring overlap • Merge the two fragments • Repeat until no more merges can be done
Overlap-layout-consensus Main entity: read Relationship between reads: overlap 1 4 7 2 5 8 3 6 9 2 3 4 5 6 7 8 9 1 ACCTGA ACCTGA AGCTGA ACCAGA 1 2 3 2 3 1 1 2 3 3 1 1 2 3 1 3 2 2
Paths through graphs and assembly • Hamiltonian circuit: visit each node (city) exactly once, returning to the start Genome
Overlap between two sequences overlap (19 bases) overhang (6 bases) …AGCCTAGACCTACAGGATGCGCGGACACGTAGCCAGGAC CAGTACTTGGATGCGCTGACACGTAGCTTATCCGGT… overhang % identity = 18/19 % = 94.7% • overlap - region of similarity between regions • overhang - un-aligned ends of the sequences • The assembler screens merges based on: • length of overlap • % identity in overlap region • maximum overhang size.
All pairs alignment • Needed by the assembler • Try all pairs – must consider ~ n2 pairs • Smarter solution: only n x coverage (e.g. 8) pairs are possible • Build a table of k-mers contained in sequences (single pass through the genome) • Generate the pairs from k-mer table (single pass through k-mer table) k-mer
RptA RptB 3 6 9 12 2 5 8 11 1 4 7 10 13 6 4 8 10 2 12 1 13 3 11 5 9 7
4 6,10 8 2 12 1 13 3 7 11 5,9 Non-repetitive overlap graph
Handling repeats • Repeat detection • pre-assembly: find fragments that belong to repeats • statistically (most existing assemblers) • repeat database (RepeatMasker) • during assembly: detect "tangles" indicative of repeats (Pevzner, Tang, Waterman 2001) • post-assembly: find repetitive regions and potential mis-assemblies. • Reputer, RepeatMasker • "unhappy" mate-pairs (too close, too far, mis-oriented) • Repeat resolution • find DNA fragments belonging to the repeat • determine correct tiling across the repeat
Statistical repeat detection Significant deviations from average coverage flagged as repeats. - frequent k-mers are ignored - “arrival” rate of reads in contigs compared with theoretical value (e.g., 800 bp reads & 8x coverage - reads "arrive" every 100 bp) Problem 1: assumption of uniform distribution of fragments - leads to false positives non-random libraries poor clonability regions Problem 2: repeats with low copy number are missed - leads to false negatives
Mis-assembled repeats excision collapsed tandem rearrangement
ribosomal RNA repeats, Ames Porton strain An assembly puzzle: contradictory data(discovered after publication) “mates” “chimeras” How do you align the green pieces?
Reference: Ames ‘ancestor’ strain Puzzle solution Ames Porton Down strain Tandem duplication
Ft Detrick Porton Down Lab C Lab D Lab B Porton Strain Florida isolate Porton 2 Porton 1 Anthrax attack strain history 1981 “Ames” isolate (Ames ancestor) 1982 … ? ? ? Plasmids cured ? Attack Strain UC Berkeley Victim 2001 1998 2001
b. anthracis SNP CS-1 GBX(g) Cut of assembly 67452 from GBX0130.contig (11 bases) Cut at ungapped consensus offset 15986 (from 1), +/- 5 positions: Cut at gapped consensus offset 16009 (from 1) 11 positions Ungapped consensus TGAATGCACAC Gapped consensus TGAATGCACAC T G A A T G C A C A C Covering reads: GBZEI27TF TGAATGCACAC 26 30 34 36 33 36 36 37 36 36 36 GBXEZ08TR TGAATGCACAC 27 30 33 35 41 37 36 23 36 36 36 GBZDA09TF TGAATGCACAC 26 18 35 31 26 20 29 19 36 36 36 Summary info: P-value (10^q) -7.9 -7.8 -10.2 -10.2 -10.0 -9.3 -10.1 -7.9 -10.8 -10.8 -10.8 Quality Class 5 3 3 3 3 3 3 3 3 3 3 Coverage depth 3 3 3 3 3 3 3 3 3 3 3 Homogeneity 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 Cut of assembly 6264 from GBA0117.contig (11 bases) Cut at ungapped consensus offset 725687 (from 1), +/- 5 positions: Cut at gapped consensus offset 730468 (from 1) 11 positions Ungapped consensus TGAATACACAC Gapped consensus TGAATACACAC T G A A T A C A C A C Covering reads: GBIFW80TF TGAATA-ACAC 34 33 15 16 13 09 00 11 11 11 15 GBICA33TR TGAATACACAC 34 35 34 35 35 34 34 36 36 36 36 GBIFQ32TR TGAATACACAC 10 13 12 18 24 13 21 12 13 14 13 GBICH40TF TGAATACACAC 36 36 36 32 32 32 32 32 31 31 36 GBICU19TR TGAATACACAC 21 30 33 18 19 24 21 11 36 10 29 Summary info: P-value (10^q) -42.8 -45.4 -45.5 -45.3 -46.7 -44.4 -46.7 -43.9 -46.8 -45.1 -42.7 Quality Class 1 1 1 1 1 1 1 1 1 1 1 Coverage depth 16 16 16 16 16 16 15 16 16 16 16 Homogeneity 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0 GBA(a) Probability of base-calling error Not shown