Download

1 / 41
420 likes | 691 Views
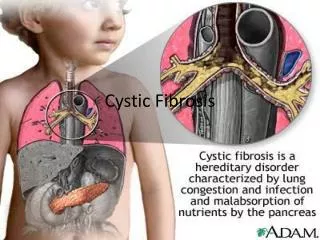
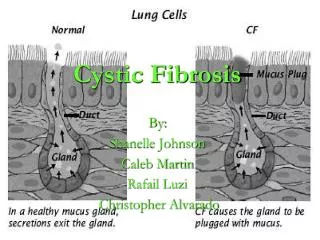
Chapter 14 Cystic Fibrosis. Figure 14-1. Cystic fibrosis. Anatomic Alterations of the Lungs. Excessive mucus production and accumulation of thick, tenacious mucus in the tracheobronchial tree Partial or total bronchial obstruction (mucus plugging) Atelectasis Hyperinflation of the alveoli.

E N D
Anatomic Alterations of the Lungs • Excessive mucus production and accumulation of thick, tenacious mucus in the tracheobronchial tree • Partial or total bronchial obstruction (mucus plugging) • Atelectasis • Hyperinflation of the alveoli
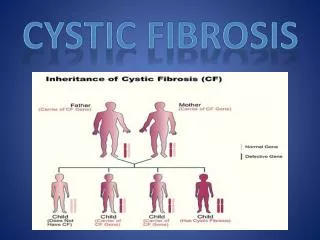
Etiology • Cystic fibrosis is an autosomal recessive gene disorder caused by mutations in a pair of genes located on chromosome 7. • Under normal conditions, every cell in the body (except the sex cells) has 46 chromosomes— • 23 pairs (one half inherited from father and the other half from mother).
Over 1000 different mutations in the gene that encodes for the cystic fibrosis transmembrane conductance regulator (CFTR) have been described. One genetic defect linked to cystic fibrosis involves the absence of three base pairs in codon 508 (ΔF508) that code for phenylalanine on chromosome 7 (band q31). This is the most common genetic mutation associated with cystic fibrosis and accounts for 70% to 75% of the cystic fibrosis patients tested. Etiology (Cont’d)
Figure 14-2. Standard Mendelian pattern of inheritance of cystic fibrosis.
Screening and Diagnosis (Cont’d) Sweat Test Immunoreactive Trypsinogen Test Stool Fecal Fat Test Nasal Potential Difference (NPD) Genetic Testing Prenatal Testing Amniocentesis Chorionic villus biopsy
Overview of the Cardiopulmonary Clinical Manifestations Associated with Cystic Fibrosis The following clinical manifestations result from the pathophysiologic mechanisms caused (or activated) by Atelectasis Bronchospasm Excessive Bronchial Secretions
Clinical Data Obtained from Laboratory Tests and Special Procedures
PaO2 and PaCO2 trends during acute alveolar hyperventilation.
PaO2 and PaCO2 trends during acute or chronic ventilatory failure.
Figure 14-4. Chest X-ray of a patient with cystic fibrosis. Note the lung overinflation, the diffuse infiltrates, and the large main pulmonary artery segment.
Figure 14-5. Cystic fibrosis. Serial chest imaging over a 26 year period showing the progressive changes of cystic fibrosis. A, At 3 years of age, the patient had right middle lobe pneumonia. B, Mild hyperinflation and bronchial wall thickening (arrow) by age 7 years. C, At age 15 years, the patient demonstrates progressive hyperinflation, bronchiectasis, and enlargement of the hila on the chest radiograph. D, Lateral chest radiograph at 29 years shows typical findings of end-stage cystic fibrosis. Note marked hyperinflation and “barrel chest” deformity, severe bronchiectasis, and tubular opacities consistent with mucous plugs.
Common Nonrespiratory Clinical Manifestations • Meconium ileus • Meconiumileus equivalent • Malnutrition and poor body development • Deficiencies of vitamins A, D, E, and K • Nasal polyps and sinusitis • Infertility (males)
General Management of Cystic Fibrosis • Patient and family education • Respiratory care treatment protocols • Oxygen therapy protocol • Bronchopulmonary Hygiene Therapy Protocol • Lung Expansion Therapy Protocol • Aerosolized Medication protocol • Mechanical Ventilation Protocol
Figure14-6. An 18-month-old female cystic fibrosis patient wearing a high-frequency chest compression (HFCC) vest (the inCourage System).
General Management of Cystic Fibrosis (Cont’d) Medication and special procedures prescribed by the physician • Xanthines • Expectorants • Antibiotics • Lung or heart/lung transplantation • Future Treatments • Some advances in gene therapy
Figure14-7. Chest x-ray of a 27-year-old man with cystic fibrosis.