Download
1 / 1
10 likes | 171 Views
Ab initio calculations of the excited states and spectra of HCl Erlendur J ónsson, Andras Bödi, Victor Wang, Ágúst Kvaran Science Institute University of Iceland.

E N D
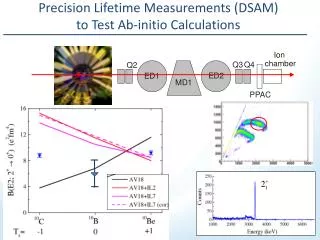
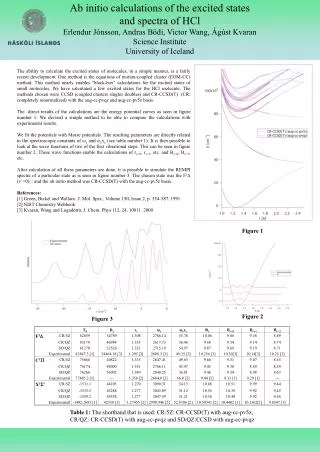
Ab initio calculations of the excited states and spectra of HCl Erlendur Jónsson, Andras Bödi, Victor Wang, Ágúst Kvaran Science Institute University of Iceland The ability to calculate the excited states of molecules, in a simple manner, is a fairly recent development. One method is the equations of motion-coupled cluster (EOM-CC) method. This method nearly enables "black-box" calculations for the excited states of small molecules. We have calculated a few excited states for the HCl molecule. The methods chosen were CCSD (coupled clusters singles doubles) and CR-CCSD(T) (CR: completely renormalized) with the aug-cc-pvqz and aug-cc-pv5z basis. The direct results of the calculations are the energy potential curves as seen in figure number 1. We devised a simple method to be able to compare the calculations with experimental results. We fit the potentials with Morse potentials. The resulting parameters are directly related to the spectroscopic constants of e and exe (see table number 1). It is then possible to look at the wave functions of two of the first vibrational steps. This can be seen in figure number 2. These wave functions enable the calculations of rv=0, rv=1, etc. and Bv=0, Bv=1, etc. After calculation of all these parameters are done, it is possible to simulate the REMPI spectra of a particular state as is seen in figure number 3. The chosen state was the F1 (v’=0) and the ab initio method was CR-CCSD(T) with the aug-cc-pv5z basis. References: [1] Green, Bickel and Wallace. J. Mol. Spec., Volume 150, Issue 2, p. 354-387. 1991 [2] NIST Chemistry Webbook [3] Kvaran, Wang and Logadóttir, J. Chem. Phys 112, 24, 10811. 2000 Figure1 Figure2 Figure3 Table 1: The shorthand that is used: CR/5Z: CR-CCSD(T) with aug-cc-pv5z, CR/QZ: CR-CCSD(T) with aug-cc-pvqz and SD/QZ:CCSD with aug-cc-pvqz