Download

1 / 44
440 likes | 875 Views
Outline. How does Aspirin work?!Structure-based rational drug designWhat is Docking?Current approaches and software packagesWhy is protein flexibility important?One step forward in flexible docking .... What Is Aspirin?. Acetosal: Route of administration: OralStructure:NOTE: Images without reference are taken from public domain (mostly Wikipedia).

E N D
1. Toward Fully Flexible Docking Bashir S. Sadjad
bssadjad@uwaterloo.ca
School of Computer Science, University of Waterloo, Canada
Simulated Biomolecular Systems, Toronto, Canada
2. Outline How does Aspirin work?!
Structure-based rational drug design
What is Docking?
Current approaches and software packages
Why is protein flexibility important?
One step forward in flexible docking ...
3. What Is Aspirin? Acetosal:
Route of administration: Oral
Structure:
NOTE: Images without reference are taken from public domain (mostly Wikipedia)
4. How does Aspirin Work? A painkiller, also used against fever
Reduce production of Prostaglandin, Thromboxane
Prostaglandin:
Prostaglandin binds to some trans-membrane proteins of spinal neuron cells causing pain.
5. How does Aspirin Work? (cont'd) Cyclooxygenase is inhibited (breaking the pathway).
Thromboxane pathway:
Many of drugs interfere in a protein function.
Aspirin effect is irreversible, we like reversible!
6. Enzymes (Review) Enzymes are great catalyzers (may speed up a reaction by 5 to 17 order of magnitude).
An explanation of how they work:
7. Enzyme Inhibition Change of shape or chemical properties of active site on an enzyme.
Example: blocking active site in HIV protease by ritonavir.
8. Outline How does Aspirin work?!
Structure-based rational drug design
What is Docking?
Current approaches and software packages
Why is protein flexibility important?
One step forward in flexible docking ...
9. How Was Aspirin Discovered? Roots can be traced back to 5th century BC, in Hippocrates notes regarding a bitter powder extracted from willow bark easing pain.
Similar references found in other ancient notes.
Willow produces salicin
which acts similarly to
aspirin in the body.
Main discovery was done in 19th century.
10. Rational Drug Design The discovery was by trial and error anyway!
How a similar drug may be discovered in 21st century?
Identify related pathways
Select target proteins
Identify active sites and allosteric sites
Try to inhibit target proteins
Inhibition may happen by ligand binding
11. Rational Drug Design (Example) Zanamivir (a ligand) used for treatment of Influenza virus.
Inhibits Neuraminidase, an enzyme on the surface of Influenza virus.
One of the first rationally designed drugs, by Biota (1989).
Marketed by GSK (1999).
12. Outline How does Aspirin work?!
Structure-based rational drug design
What is Docking?
Current approaches and software packages
Why is protein flexibility important?
One step forward in flexible docking ...
13. High Throughput Screening (HTS) Once the target protein is purified a library of ligands might be tested against it.
The size of a practical library is in 105 � 106 range.
Ligands active against the target protein can be selected by automated mechanisms.
This requires significant resources including expensive labs.
14. Virtual HTS How if we can predict the HTS result by a computer program?
This is Virtual High Throughput Screening.
One way to do this is by simulating the binding process using properties of involved molecules.
Free Energy of Binding determines the affinity.
15. Free Energy Each conformation and binding mode has a specific free energy:
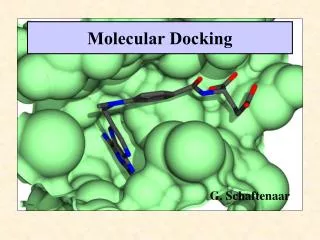
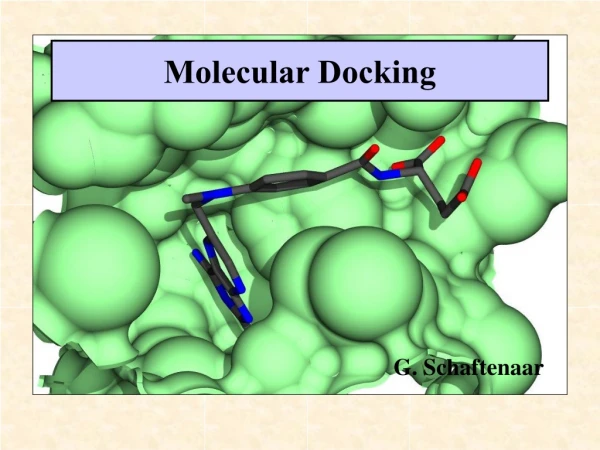
16. Docking Determine the best binding mode:
An approximation of free energy is used (scoring function).
The search engine finds the minimum of the scoring function.
Carbonic anhydrase and a bound ligand
17. Outline How does Aspirin work?!
Structure-based rational drug design
What is Docking?
Current approaches and software packages
Why is protein flexibility important?
One step forward in flexible docking ...
18. Classification Criteria Type of scoring function:
Force-field based
Empirical
Statistical
Search method:
Systematic
Stochastic
Degree of ligand-protein flexibility
19. Scoring Approximating reality: a bad approximation won't work even with the best search method.
A general form for an empirical scoring function:
20. Search Stochastic and heuristic
MCDOCK (simulated annealing)
AutoDock2 (simulated annealing)
GOLD (genetic algorithm)
Directed
FlexX (incremental construction)
eHiTS (exhaustive search)
Combined
Glide (systematic pose gen. + stochastic optimization)
21. eHiTS eHiTS approach [4]:
Ligand fragmentation
Fragment rigid-dock
Fragment matching
Ligand reconstruction
Local optimization
22. Example (Rigid Docking) Each fragment is docked in cavity.
For sufficient accuracy a fine sampling should be done.
23. Example (Matching) All fragments are scored.
A diverse set of matching poses with high scores are selected.
A full ligand pose is generated from each matching set.
24. Outline How does Aspirin work?!
Structure-based rational drug design
What is Docking?
Current approaches and software packages
Why is protein flexibility important?
One step forward in flexible docking ...
25. Protein Structure and Ligand Binding Protein structure may significantly be changed by ligand binding.
Calmodulin (a calcium-binding protein):
Movie: http://molmovdb.org/cgi-bin/morph.cgi?ID=78252-5656
26. The Allosteric Effect Binding of ligands may regulate the protein function.
Example: binding of oxygen and carbon-dioxide to hemoglobin:
27. Binding Site Flexibility Ligand binding changes the binding site of the protein. This is called induced fit.
In many of protein-ligand complexes in PDB, the cavity surrounds the ligand with a small open part.
Rigid treating of binding site (as done by most docking programs), makes binding energy prediction difficult.
28. Example (Ligand Binding) Conformational change at the binding site of Renin.
29. Example (Cavity Closure) L-Arabinose-binding protein complexed with L-Arabinose. (PDB: 1ABE)
30. A Note on Structure-Function Assumption Amino Acid � Structure � Function assumption.
Consider a highly hydrophilic protein sequence, is it folded in water? Does it have any functions?
Indeed it is not in a single folded state but it can be functional! There are functional intrinsically unstructured proteins.
They may fulfil different tasks and have different fold for each task.
31. Example (Unstructured) The pKID domain of CREB protein, complexed with KIX domain of CREB-binding protein.
32. Example (Structural Change) The TAZ1 domain of CREB-binding protein complexed with two different domains.
33. Outline How does Aspirin work?!
Structure-based rational drug design
What is Docking?
Current approaches and software packages
Why is protein flexibility important?
One step forward in flexible docking ...
34. Truly Flexible Docking A truly flexible docking application is in fact a folding program!
eHiTS is an ab-initio method: folding complexity
Different types of protein mobility:
Movement of large domains
Multiple conformations observed in a few residues
35. Movement of Domains Patterns of domain movement:
Ribose-binding protein movie (2DRI, 1URP):
http://molmovdb.org/cgi-bin/morph.cgi?ID=645772-17065
36. Conformations of a Few Residues Acetylcholinesterase (PDB: 2ACE, 1EVE, 1VOT, 1ACL)
37. Truly Flexible Docking A truly flexible docking application is in fact a folding program!
eHiTS is an ab-initio method: folding complexity
Different types of protein mobility:
Movement of large domains
Multiple conformations observed in a few residues (to be addressed in first step)
38. Binding Site Side-Chains Modeling side-chain flexibility of binding site residues in eHiTS.
First the candidate chains should be selected.
Solvent exposed?
More statistics
39. Binding Site Side-Chains (cont'd) Same technique of fragmentation can be applied to side-chains.
Rigid docking and pose matching with the backbone constraints.
40. The Problem Size (Difficulty) Run statistics for a set of 20 PDB codes (all numbers are averages):
# rigid fragments: 3.05
# poses tried in RD: 60 million
# poses accepted in RD: 493,354
Best Match RMSD: 0.60 A
Best Match Found: 1.01A
(NOTE: Finding the best match is NP-hard for a general scoring function.)
41. Pose Match Example Closest match for an HIV protease inhibitor (1AAQ):
42. Training eHiTS uses a statistical scoring function.
Training is done by known structures.
Pose Match specific training is done by linear programming modeling and using CLP package.
For receptor flexibility modeling we can either:
Generate receptor decoys
Use PDB complexes with same receptor
43. Goals and Previous Works Induced fit modeling in Glide [5]:
Docking into rigid receptor using softened scoring func.
Receptor active site sampling
Complex optimization (minor backbone flexibility)
Differences with our approach:
Simultaneous handling of ligand/receptor flexibility
Same scoring function (no softened version)
The set of cross-docking data can be used for training and benchmarking.
44. Selected References S. J. Teague, Implications of Protein Flexibility for Drug Discovery, Nature Reviews (Drug Discovery), vol. 2, pp. 527-541, 2003.
D. B. Kitchen, H. Decornez, J. R. Furr, J. Bajorath, Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications, Nature Reviews (Drug Discovery), vol. 3, pp. 935-949, 2004.
H. J. Dyson, P. E. Wright, Intrinsically Unstructured Proteins and Their Function, Nature Reviews (Molecular Cell Biology), vol. 6, pp. 197-208, 2005.
Z. Zsoldos, D. Reid, A. Simon, B. S. Sadjad, A. P. Johnson, eHiTS: A New Fast, Exhaustive Flexible Ligand Docking System, J. of Mol. Graphics and Modeling, (to appear � available online).
W. Sherman, T. Day, M. P. Jacobson, R. A. Friesner, R. Farid, Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects, J. Med. Chem., vol. 49, pp. 534-553, 2006.
45. Questions?