Download

1 / 38
390 likes | 547 Views
Diversité des transcrits: Les sites de polyadenylation « intergéniques ». Daniel Gautheret, 2005 INSERM ERM206 Université de la Méditerranée. Annotation des génomes. Humain/souris 2005: 22000 gènes annotés Est-on proche de la fin?

E N D
Diversité des transcrits:Les sites de polyadenylation « intergéniques » Daniel Gautheret, 2005 INSERM ERM206 Université de la Méditerranée
Annotation des génomes • Humain/souris 2005: 22000 gènes annotés • Est-on proche de la fin? • Si peu de différence avec la mouche (15000 gènes) ou C. elegans (18000) ? Annotation Ensembl
Les EST (expressed sequence tags): l’outil No 1 pour l’annotation des génomes
Hier: clustering d’ESTs pour reconstruire les transcrits (avant 2001: génomes vertébrés non disponibles) Cluster d’ESTs Transcrit reconstruit
Aujourd’hui: Alignements EST/génome exons introns
Alignement EST/génome Skipped exon
Epissage alternatif 3 gènes de détermination du sexe chez la drosophile, épissés différemment selon le sexe de l'individu:
Epissage alternatif via EST • Nombreux travaux réalisés • Sociétés créées exclusivement sur le thème (par ex. Compugen http://www.cgen.com/) • Ex: Modrek B, Resch A, Grasso C, Lee C.Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res 2001 Jul 1;29(13):2850-9 :
ASAP, AltExtron, ASD, SpliceNest… ASAP interface (Lee et al. 2003)
Travaux récents sur la diversité du transcriptome • Tiling array • Full-length cDNA
Le Tiling Array • Rosetta: technologie de spottage d’oligonuléotides par jet d’encre (inkjet)
Gène bien caractérisé: confirmation des exons « refseq » et apparition de nouvelles régions transcrites (ici correspondant à un cDNA déjà observé)
Apparition de nouveaux gènes dans régions intergéniques sans EST ni prédiction bioinformatique.
Que trouve-t-on? • Nouveaux gènes codants • >2000 transcrits avec ORF non existant dans Ensembl • Nouveaux gènes non-codants • >10000 transcrits • Transcrits antisens • >5000 gènes ont transcription antisens • Isoformes de gènes connus • Au moins 40% à 60% des gènes selon auteurs • Comprend également extensions (parfaois très longues) en 3’ • Artefacts • biologiques: « bruit » transcriptionnel • expérimentaux: hybridation non-spécifique, contamination génomique…
Tiling 2 • Technologie: Affymetrix • 25-mères espacés de 5bp • polyA+ et polyA- • Noyau et cytoplasme • 8 lignées cellulaires différentes Science, 2005
Etat de polyadenylation • Parmi tous les transcrits observés: • 19% polyA+ • 44% polyA- • 37% polyA+ et polyA- • Donc la moitié du transcriptome humain est polyA- • Important, car les transcrits qu’on regardait jusqu’à présent n’étaient que les polyA+!
Position des transcrits • 60% des loci exprimés présentent des évidences de transcription sur 2 brins • Beaucoup de transcription dans l’intergénique: • 50% des polyA- cytoplasmiques et 25% des polyA- nucléaires sont intergéniques • 41% des polyA+ nucléaires sont intergéniques
100,000 Full length cDNAs + 1M « CAGE » (sortes de SAGE en 5’) Full length cDNAs Science, 2005
Utilité des full-lengths • Etude des promoteurs (impossible avec les EST) • TSS alternatifs, exons 5’ alternatifs
Transcrits et Unités de Transcription • TU: Transcription Unit. mRNAs sharing at least 1 nt and with same location and orientation
Conclusion projet « FANTOM3 » • 32000 transcrits non-codants • 16000 nouveaux transcripts codants • 5000 nouvelles protéines • La majorité du génome est transcrit sur les deux brins • La grande diversité des transcrits dans chaque TU soulève le problème de la précision des puces ADN conventionnelles, dans lesquelles chaque sonde hybride différents transcrits
Les besoins actuels • Intégration initiation+épissage+transcription • Caractérisation des nouveaux transcrits • Etude fonctionnelle (domaines, etc.) • Conservation • Validation expérimentale • Tissu-spécificité The Alternative Transcript Diversity Project (ATD), 6e PCRD
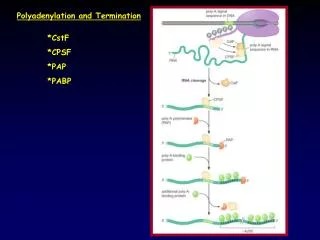
La polyadénylation alternative Cleavage + polyA-addition site Pre-mRNA PolyAdenylation Signal Coding sequence 3' UTR AAUAAA CA GUUGU AUUAAA CA GUUGU To 5' end AAAAAAAAA... AAAAAAAAA... mRNA alternative transcripts
PAS Discovery through EST/mRNA Alignment mRNA or EST-contig 5’ESTs ESTs 3’ESTs First observation in 1998: 189 cases of alternative polyadenylation 2000: 1000 cases Gautheret et al. (1998) Genome Res. 8, 524 Beaudoing et al. (2000) Genome Res. 10, 1001
EST-based PAS Map 2005 human mouse chicken Tot PAS 66,600 52,300 3,500 PAS <3K from ENSEMBL Gene 31,000 27,900 2,900 PAS >10K from ENSEMBL Gene 29,000 19,400 300 Genes with no PAS 30% 36% 80% Genes with 2 or more PAS* 50% 43% 7% *relative to all genes with 1+ PAS -> 54% human, 32% mouse (accept multiple sites for 1 signal, but refseq UTR only)
Intergenic PAS? • « Intergenic » PAS are made of • False positives • Long range PAS • PAS from novel genes stop stop
What is the actual reach of 3’UTRs? • Textbook « Human Molecular Genetics 2 » (1999): • 3′ UTR Average of about 0.6 kb (see Zhang, 1998) but this is likely to be an underestimate because of underreporting of genes with long 3′ UTRs • Untranslated Regions of mRNA (Mignone et al. 2003) :
Several recent papers mentioning distal PAS • All rely on EST sampling, but: • Require alignment on refseq gene/fl cdDNA or overlapping ESTs • Cannot assess all long range PAS
Site distribution ? 9kb Ratio 5’ supported # sites in 500nt window Distance from STOP • 5’ support is significant up to 9kb past Stop codon • May be limited by cloning capacity (few inserts larger than 6kb)
cDNA 3’EST 5’EST Position of 5’ EST in transcript start stop Relative position of 5’ EST (% of transcript) Distance of 3’ EST from STOP • As distance from Stop increases, 5’ EST are located closer to 3’ end • After >6kb, most 5’ EST are « out of gene »
Classification Trees for PAS prediction • 8 Variables: • canonical/non canonical signal • Erpin score for downstream region • #supporting ESTs • PolyA/T in ESTs • ESTs overlap previous site • Full-length cDNA • Single/multiple EST libraries • 5’EST from same clone mapping within gene Class 1 Class 2 stop 5kb 10kb 15kb 20kb
Classification tree + Voting Procedure -> class 1 / class 2
Importance of variables • Canonical/non canonical • Erpin score • #supporting ESTs • polyA/T in ESTs • Overlap with previous site • Full-length cDNA • Single/multiple EST libraries • 5’EST from same clone All variables No 5’ EST
Average P-value vs Distance @20k: ave P-value=0.21 20% have P-value >0.5 Relative position of 5’ EST (% of transcript) 13kb Distance from STOP • 20% of long distance sites correspond to bona fide PAS
Conclusions • P-values for polyA sites • 6000 Novel polyA+ genes in intergenic • Average 3’ UTR longer than measured based on 5’ EST mapping: around 2kb mean. • Consequence for ncRNA search in UTRs
Equipe • polyA group @TAGC • Matthieu Legendre • William Ritchie • Takeshi Ara • Fabrice Lopez • Arbres de classification: • Badih Ghattas, Département de Mathématiques, Aix-Marseille II