Download
1 / 43
430 likes | 717 Views
Challenges to therapy for X-linked adrenoleukodystrophy. Nancy Braverman, MS, MD McGIll University-MCH-RI. March 11 2010 HGEN 171-575. Peroxisomes originate from ER membranes and by fission of existing peroxisomes. NEXT >>. Click to view animation >>.

E N D
Challenges to therapy for X-linked adrenoleukodystrophy Nancy Braverman, MS, MD McGIll University-MCH-RI March 11 2010 HGEN 171-575
Peroxisomes originate from ER membranes and by fission of existing peroxisomes NEXT >> Click to view animation >> adapted from Annu Rev Genet. 2000;34:623-652. Sacksteder KA, Gould SJ.
Role of peroxins in matrix protein import Click to view animation >> Gould, Raymond, Valle.In: Metab & Molec Basis of Inh Dis. Ch 129 p. 3190.
The 3 major metabolic pathways in peroxisomes mitochondria
Contain Peroxisome Targeting Sequences (PTS) Imported as oligomers/fully assembled proteins Can have dual localizations in mitochondria, cytosol Properties of peroxisomal matrix proteins PTS1 PTS2 -SKL R/KLX5Q/HL -SKL N - terminal (-R/KLX5 Q/HL-) Presequence cleaved internally 3 enzymes only: Thiolase, PhyH, AGPS Receptor is PEX7 C - terminal (-SKL) Most matrix proteins Receptor is PEX5
Genetic disorders of peroxisomes • Multiple enzyme deficiencies: Peroxisomal Biogenesis Disorders (PBD) • Zellweger spectrum disorder (ZSD) (~1/60,000) • Rhizomelic chondrodysplasia punctata spectrum (RCDP)(~1/100,000) • Single enzyme deficiencies • X-linked adrenoleukodystrophy (X-ALD) (~1/20,000) • 3-methyl-CoA racemase deficiency • Adult Refsum disease • Hyperoxaluria Type I (Primary Hyperoxaluria)
Develop therapies targeted to the metabolic defects Phytanic acid restriction Reduction in VLCFA dietary reduction enhance VLCFA omega oxidation reduce VLCFA synthesis Supplementation with DHA, bile acids, plasmalogens A--------->B
Develop therapies targeted to the molecular defects • Enhance activity of a defective PEX protein- improve protein folding • Bypass the need for a specific PEX protein- upregulate a partner PEX protein • Induce peroxisome proliferation • Enzyme/PEX protein replacement therapy ? • Liver/bone marrow stem cell transplant ? • Gene therapy ? • Manipulate the intestinal microbiome?
PBD phenotypes correlate with: • Severity of the biochemical defects • Severity of import defect, peroxisome number & size • Predicted effect of the PEX gene mutation on protein function • nonsense, frameshift, deletion alleles • no residual protein function • missense alleles some residual protein function
Mild PBD, PEX1-p.G843D/G843D Pxmembprot CATALASE PTS1/PTS2 37oC 30oC
Conformational changes of p97 AAA ATPase during its ATPase cycle Bind and hydrolyze ATP generating chemical energy that is converted into motion of the molecule. Motion used to pull PEX5 out of the membrane for another round of import
How do we improve protein folding? • Lower temperature • Chaperone (protein or drug) • Nonspecific chemical chaperone • Pharmacologic chaperone Enzyme substrate Protein/enzyme inhibitor -protein kinase and kinase inhibitor Vitamin cofactor
Intracellular distribution of AGT, a protein with an N-terminal MTS & C-terminal PTS1
Can we manipulate peroxisome protein targeting for therapy? Cell penetrating peptides: Short sequence domains from known proteins that are able to translocate across the plasma membrane HIV-TAT, penetrin, octa-arginine Fuse these domains to: PTS1/PTS2 matrix protein PEX proteins?
Better understanding of the disease pathophysiology may reveal novel targets for therapy: mouse models • Selective inactivation of Pex5 gene in neural cells (conditional gene targeting)
X-linked adrenoleukodystrophy (X-ALD) • Defect in adrenoleukodystrophy protein (ALDP) encoded by the ABCD1 gene) • Mapped to Xq28 • Over 200 mutations known, most result in no detectable ALDP protein • Incidence ~ 1/20,000 • All ethnic groups • Reduced peroxisomal VLCFA oxidation
Clinical picture in a child with the cerebral form • Medical history • 8-year old previously healthy, normal male • Attention deficit/hyperactivity apparent within the past year • Performing poorly in 2nd grade • Recently began to run clumsily and to walk stiffly • No recent illnesses • No medications
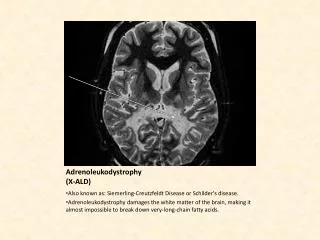
Rapid deterioration in the X-ALD cerebral form Deterioration in writing over a 4 month period Brain MRI – white matter disease Dec 29, 1989 Mar 5, 1990 May 3, 1990
X-ALD: defective peroxisomal β-oxidation Click to view animation >>
Childhood cerebral form ~35% Onset - ~6-12 yrs (survival: several years) 90% with adrenal insufficiency Adrenomyeloneuropathy (AMN) ~50% Spastic paraparesis and sphincter dysfunction Onset - ~2nd-5th decade (survival: decades) 2/3 with adrenal insufficiency Other phenotypes ~15% adrenal insufficiency only Adult-onset cerebral involvement - dementia Female heterozygotes- 50% with milder AMN symptoms Multiple phenotypes of X-ALD
X-ALD pedigree: ABCD1-c.1801delAG Adrenal disease 0 Spastic paraparesis (AMN)
Lack of genotype-phenotype correlation in X-ALD • All clinical phenotypes are observed in the same nuclear family • Deletion mutations are associated with severe and mild phenotypes • Monozygotic twins are reported with different phenotypes
X-ALD treatment: dietary changes • Dietary therapy • Restriction of dietary VLCFA intake • Lorenzo’s oil- 4:1 mix • Glycerol trioleate (C18:1) • Glycerol trierucate (C22:1) • Lowers plasma C26:0 and C24:0 levels • Reduces, but does note eliminate the risk for the childhood leukodystrophy phenotype
X-ALD: other approaches to therapy • Cholesterol lowering drugs (Lovastatin) • Increase omega oxidation of VLCFA • Reduce endogenous elongation of VLCFA • Induce expression of partner proteins (the anticonvulsant, valproate, induces ABCD2 expression)
X-ALD treatment: allogenic bone marrow transplantation (from donor) • Colonisation of the brain by cells of the monocyte-macrophage system (become microglial cells) provides the rationale for the use of BMT in X-ALD • Assymptomatic boys are put on LO and followed closely for cerebral involvement • At first signs of cerebral disease, a transplant is recommended
X-ALD pedigree Adrenal disease 5 months 0 Spastic paraparesis (AMN) ? X-ALD
Environmental factors and candidate modifier gene(s) in X-ALD • Environmental factor as the initial trigger of cerebral inflammation eg viral infection • Genetic segregation analysis supports the role of at least one autosomal dominant modifier gene • Polymorphisms in ABCD2 (ALDPR), ABCD3 (PMP70), ABCD4 (PMP70R) • Polymorphisms in genes encoding inflammatory proteins • Polymorphisms in elongation of VLCFA (ELOV1)
Identifying modifier genes in X-ALD by SNP association studies ABCD2 polymorphisms and clinical phenotypes showed an even allele distribution in different X-ALD phenotypes and controls Genes involved in methionine metabolism: weak association with a polymporphism in Tc2
ABCD1 null mouse model • Has elevation of VLCFAs • Does not develop cerebral disease • Older mice develop axonal degeneration
A comeback for gene therapy: ex vivo gene correction 20 months ALD patient Reduction in VLCFA Gene-corrected HSCs HIV-based vector with therapeutic ABCD1 gene Progeny of gene-corrected HSCs distribute throughout the body
Lentiviral vectors • Retroviruses, adenoviruses, adeno-associated viruses and lentiviruses are used in genetic engineering. • The most commonly used rLV vector is based on the human immunodeficiency virus 1 (HIV-1)
Recombinant lentiviral gene therapy vectors vs. other retroviral vectors • TransduceHSCs more efficiently • Self-inactivating long terminal repeats do not promote transcription, thus reducing the risk of mutagenesis and leukemia • Infects non-dividing cells (neurons)
ABCD1 gene transfer with lentiviral vectors: preclinical studies • Cell model: Transduced HsALDP- ,CD34+ cells (pluripotent HSC)biochemical correction of derived monocytes/macrophages • Whole animal model: Transduced MmALDP-, CD34+ cells into X-ALD mice replacement of 25% brain microglial cells, but mice do not devlop a leukodystrophy so cannot tell if treatment is clinically effective
Hematopoetic stem cell lineages Monocyte derived cells include brain microglial cells
2 patients (7 yo) with cerebral disease with no matched donor • CD34+ cells isolated, infected with HIV-1 lentiviral vector-ABCD1 • Patients underwent bone marrow ablation, in order to repopulate the bone marrow with the engineered cells • Transduced cells were re-infused into the patient without complications
Patient results: did it work? A-C. Expression of ALDP in PBMCs by IF using an ALDP antibody (CD14-moncytes, CD15 granulocytes, CD3 T lymphocytes, CD19 B lymphocytes) D. Plasma C26:0/C22:0 levels, grey band is normal values Long term expression in PBMCs, continue expression in CD34+ cells
Analysis of LV insertion sites • Showed multitude of insertion sites without clonal dominance
Neurological outcomes (A) Is patient 1 and (B) is pateint 2before and after gene therapy (C) Is in an untreated 8-year-old ALD patient showing the progression of cerebral demyelinating lesions
Conclusions: is therapeutic efficacy≅ allogenic BMT? • Clinical outcome similar so far • Data support HSC engraftment with capacity to repopulate multiple hematopoetic lineages • Up to 14% of cells in each lineage expressed ALDP in contrast to 100% in allogenic BMT • However, ABCD1 gene was overexpressed by its promoter, and this may have helped to reduce VLCFA to equivalent levels