Download
1 / 1
10 likes | 90 Views
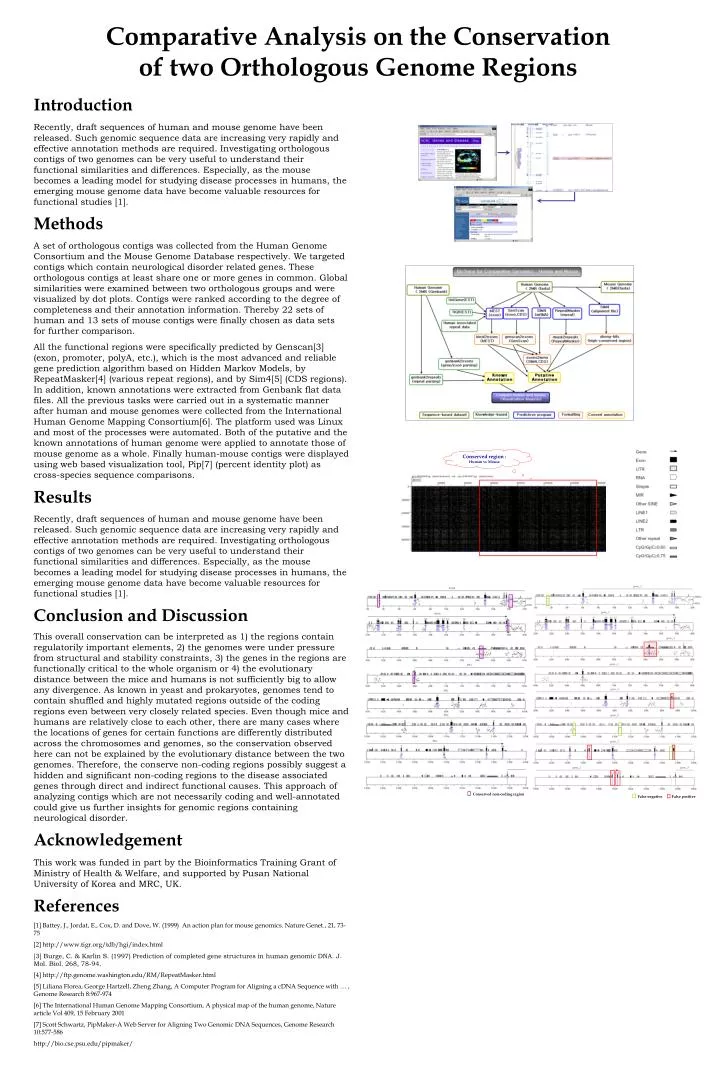
Conserved non-coding region. False positive. False negative. Conserved region : Human vs Mouse. Introduction.

E N D
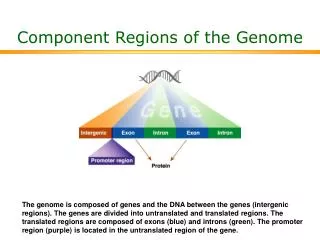
Conserved non-coding region False positive False negative Conserved region : Human vs Mouse Introduction Recently, draft sequences of human and mouse genome have been released. Such genomic sequence data are increasing very rapidly and effective annotation methods are required. Investigating orthologous contigs of two genomes can be very useful to understand their functional similarities and differences. Especially, as the mouse becomes a leading model for studying disease processes in humans, the emerging mouse genome data have become valuable resources for functional studies [1]. Methods A set of orthologous contigs was collected from the Human Genome Consortium and the Mouse Genome Database respectively. We targeted contigs which contain neurological disorder related genes. These orthologous contigs at least share one or more genes in common. Global similarities were examined between two orthologous groups and were visualized by dot plots. Contigs were ranked according to the degree of completeness and their annotation information. Thereby 22 sets of human and 13 sets of mouse contigs were finally chosen as data sets for further comparison. All the functional regions were specifically predicted by Genscan[3] (exon, promoter, polyA, etc.), which is the most advanced and reliable gene prediction algorithm based on Hidden Markov Models, by RepeatMasker[4] (various repeat regions), and by Sim4[5] (CDS regions). In addition, known annotations were extracted from Genbank flat data files. All the previous tasks were carried out in a systematic manner after human and mouse genomes were collected from the International Human Genome Mapping Consortium[6]. The platform used was Linux and most of the processes were automated. Both of the putative and the known annotations of human genome were applied to annotate those of mouse genome as a whole. Finally human-mouse contigs were displayed using web based visualization tool, Pip[7] (percent identity plot) as cross-species sequence comparisons. Results Recently, draft sequences of human and mouse genome have been released. Such genomic sequence data are increasing very rapidly and effective annotation methods are required. Investigating orthologous contigs of two genomes can be very useful to understand their functional similarities and differences. Especially, as the mouse becomes a leading model for studying disease processes in humans, the emerging mouse genome data have become valuable resources for functional studies [1]. Conclusion and Discussion This overall conservation can be interpreted as 1) the regions contain regulatorily important elements, 2) the genomes were under pressure from structural and stability constraints, 3) the genes in the regions are functionally critical to the whole organism or 4) the evolutionary distance between the mice and humans is not sufficiently big to allow any divergence. As known in yeast and prokaryotes, genomes tend to contain shuffled and highly mutated regions outside of the coding regions even between very closely related species. Even though mice and humans are relatively close to each other, there are many cases where the locations of genes for certain functions are differently distributed across the chromosomes and genomes, so the conservation observed here can not be explained by the evolutionary distance between the two genomes. Therefore, the conserve non-coding regions possibly suggest a hidden and significant non-coding regions to the disease associated genes through direct and indirect functional causes. This approach of analyzing contigs which are not necessarily coding and well-annotated could give us further insights for genomic regions containing neurological disorder. Acknowledgement This work was funded in part by the Bioinformatics Training Grant of Ministry of Health & Welfare, and supported by Pusan National University of Korea and MRC, UK. References [1] Battey, J., Jordat, E., Cox, D. and Dove, W. (1999) An action plan for mouse genomics. Nature Genet., 21, 73-75 [2] http://www.tigr.org/tdb/hgi/index.html [3] Burge, C. & Karlin S. (1997) Prediction of completed gene structures in human genomic DNA. J. Mol. Biol. 268, 78-94. [4] http://ftp.genome.washington.edu/RM/RepeatMasker.html [5] Liliana Florea, George Hartzell, Zheng Zhang, A Computer Program for Aligning a cDNA Sequence with … , Genome Research 8:967-974 [6] The International Human Genome Mapping Consortium, A physical map of the human genome, Nature article Vol 409, 15 February 2001 [7] Scott Schwartz, PipMaker-A Web Server for Aligning Two Genomic DNA Sequences, Genome Research 10:577-586 http://bio.cse.psu.edu/pipmaker/ Comparative Analysis on the Conservation of two Orthologous Genome Regions