
Classification of Catalyst Systems
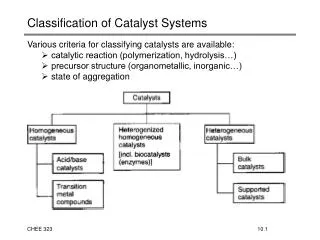
Classification of Catalyst Systems. Various criteria for classifying catalysts are available: catalytic reaction (polymerization, hydrolysis…) precursor structure (organometallic, inorganic…) state of aggregation. Homogeneous Acid-Base Catalysis of Organic Reactions.

Classification of Catalyst Systems
E N D
Presentation Transcript
Classification of Catalyst Systems • Various criteria for classifying catalysts are available: • catalytic reaction (polymerization, hydrolysis…) • precursor structure (organometallic, inorganic…) • state of aggregation
Homogeneous Acid-Base Catalysis of Organic Reactions • Catalysis of substitution, elimination and addition reactions by acids and bases is covered in CHEM 245/345, and hence, will not be a major subject of the course. • In general, acid catalysis (H+, AlCl3, etc.) operates by converting a creating a more reactive electrophile and/or converting the leaving group into a better one. • Base catalysis (HO-, amines, etc.) operates deprotonating the nucleophile to improve its reactivity
Catalysis by Homogeneous Organometallic Complexes • The study of catalysis by organometallic complexes can be justified by several reasons: • Many industrial processes exploit the remarkable selectivity and activity that homogeneous systems can provide. • Reactions catalyzed by homogeneous organometallic complexes are amongst the best understood systems. Careful study of these isolable(?) and identifiable(?) complexes helps us understand more complex systems. • Research into cluster complexes has the potential to bridge homogeneous transition metal complex chemistry with the chemistry of supported metals (heterogeneous catalysts). • In the field of catalysis, we are closest to being able to “design” organometallic catalysts for specific applications than we are for any other branch of chemistry.
OM Catalysis Examples: Controlled Isomerization • Ethylidene-norbornene is an important monomer in the synthesis of curable elastomers. A key step in the synthesis is the selective isomerization of the vinyl group to an internal position: • Three chiral centres are required to produce the characteristic • menthol odor and local • anesthetic effects. • Enantioselective • isomerization to produce • the allylic amine 20 is • required, which is done • with an organophosphine • complex of rhodium. The • final hydrogenation is • relatively easy.
OM Catalysis Examples: Asymmetric Hydrogenation • A potential synthesis of Naxopren employs an enantioselective hydrogenation step using a chiral Ru(II) chloride complex. The S-isomer is an anti-inflammatory, the R-isomer is a liver toxin.
OM Catalysis Examples: HCN Addition to Dienes • Adiponitrile is an intermediate in the nylon-6,6 process. It is generated by a catalyzed addition of HCN to butadiene: • The required addition and isomerization steps are catalyzed by a nickel catalyst, through reactions of the type:
OM Catalysis Examples: Carbomethylation of Butadiene • Adipic acid is another intermediate in the nylon process that requires organometallic catalyzed reactions. In this case, a cobalt carbonyl complex mediates the addition of CO and CH3OH to butadiene:
Bonding and Structure in Transition Metal Complexes • Organo-transition metal catalyzed • reactions are treated as • sequences of elementary • reactions. • Knowledge of tm-complex • structure is therefore important • to the study of catalytic • processes. • We will examine: • 1. Ligand bonding modes • 2. Coordination number and geometry • 3. Metal oxidation state and effective atomic number of a complex.
Bonding and Structure in Transition Metal Complexes • Much of the unique chemistry of transition metal complexes stems from partially filled d orbitals of accessible energy. • The 3d, 4s and 4p orbitals become more stable as the effective atomic number increases. Toward the middle of the series, the 3d orbitals are of lowest energy, and it is often assumed that valence electrons occupy the 3d shell.
Theories of Bonding in Inorganic Complexes • The chemistry of catalytic materials is essentially the chemistry of their complexes. A number of theories of coordination interactions have been develop, most of which are incomplete: • valence bond theory • simple electrostatic theory • ligand (crystal) field theory • molecular orbital theory. • These models of coordination compounds have been presented in fundamental Inorganic Chemistry courses. • CHEE 323 is not a basic chemistry course, but one which tries to elevate basic science to engineering practice. • Review your inorganic texts/notes regarding ligand structure and bonding theory
Classical Coordination Complexes • Classical coordination complexes are generated by those ligands that donate an electron pair. • NH3, CH3CN and H2O are ligands capable of donation of a lone pair, thereby acting as Lewis bases. • These ligands form compounds with all types of Lewis acids, metal ions or molecules. • The bond formed has rotational symmetry about the M-L axis, and is therefore called a s bond. • Non-classical ligands can accept electron density from the metal centre. Differences between tertiary amine (:NR3) complexes and trialkyl phosphine (:PR3) complexes result.
Metal Halides and Hydrides • M-X and M-H bonds are viewed from the electron-donor/electron-acceptor point of view as ionic complexes. • Electrons comprising metal-halide bonds are formally assigned to the halogen, so we speak of Cl-, Br- and I- as ligands. • Charge-radius ratio, polarizability and the ability to engage in back-bonding are important factors in M-X stability • There are many examples of transition metal halides. • The reactions of metal-hydrides are relevant to a great many catalytic reactions (isomerization, hydrogenation, hydroformylation, polymerization). • Although formally called hydrides and assigned a charge of minus 1 (as per halides), M-H bonds can range from hydridic (formally H-) to acidic (formally H+), depending on the environment about the metal.
Metal-alkyl complexes • Metal-alkyl bonds are reasonably strong (100-200 kJ/mole), but considerably weaker than M-F or M-Cl bonds (300-400 kJ/mole). • Examples: • From the perspective of catalysis, metal-alkyl complexes are important intermediates in the oligomerization, hydrogenation and hydroformylation of olefins. • While not a weak bond, M-C complexes readily undergo b-hydride elimination, which limits the degree of olefin polymerization in many systems.
Metal-carbonyl Complexes: p-acid Ligands • Carbonyl complexes are important catalytic intermediates, and CO often serves as a “neutral” ligand in support of catalytic centres. • CO donates an electron pair to the metal to generate a s-bond, but can accept electron density from the metal, into its p* orbital. • This second orbital overlap is called p back-bonding • The most important energetic component of the bonding is the ligand to metal s donation, but the back-bonding component assumes greater importance as the electron density about the metal increases
Metal-phosphine Complexes • The non-bonding electron pair of phoshines are strongly basic. Donation to generate a s bond is favoured, especially for metals in high oxidation states. • An important characteristic of phosphines is the cone angle, which defines steric bulk: • P(i-Pr3) 160o • PCy3 170o • PMe3 118o • PPh3 145o
Metal-phosphine Complexes • Phosphines differ not only in steric bulk, but in their electron donating capacity. • Consider fac-Mo(CO)3L3 which demonstrates two CO stretching resonances in the mid-infrared frequency range. • Variation of phosphine ligand (L) affects the • carbonyl stretching frequency in a very specific • manner. • L nCO cm-1 • PPh3 1934 1835 • PClPh2 1977 1885 • PCl2Ph 2016 1943 • PCl3 2024 1991 • As the donating capacity of the phosphine increases, greater electron density resides at the metal centre, which weakens the CO bond through p back bonding.
Metal-olefin Complexes • Olefins are capable of two-electron donation of the bonding p orbital electrons when oriented as shown: • The ethylene-metal bond has a component • of s symmetry corresponding to electron • donation. Overlap of a filled hybridized • 5d6p orbital with the antibonding p* orbital of the olefin generates a bond component of p symmetry.
Other p-donor Ligands: Allyl complexes • An allyl anion can act as a four electron p donor ligand by virtue of its delocalized structure: • The result is h3 coordination, where all three carbons are engaged with the metal centre. All C-C bonds are equivalent. • Reaction of allyl Grignard reagents with metal halides is a common method of synthesis. • These ligands are not of great importance to catalysis, but serve as an introduction to butadiene and cyclic diene coordination.
Other p-donor Ligands: Diene complexes • Butadiene and substituted butadienes can be trated as four electron donors. Their conjugated structure can be described in terms of the following valence bond structures: • leading to two contributing structures for metal complexes: • Butadiene complexes can be prepared by direct reaction between the ligand and a metal complex:
Other p-donor Ligands: h5-C5H5 Complexes • Dicyclopentadienyl complexes, commonly referred to as metallocenes, are an important class of polymerization catalysts. • Ligands possessing aromaticity are common: • Cp ligands are penta-hapto ligands, • donating a total of 6 electrons to • the metal and are assigned a formal • charge of -1. • Because the ligand has olefinic • character (-ene) and radical (-yl) • functionality, it is named as an • -enyl.
p-Donor Ligands: Nomenclature • Ligands capable of bonding to a metal with varying numbers of p electrons are described by hn nomenclature. • n denotes the number of carbons bonded to the metal • h is from the Greek prefix hapto (derived from haptein “to fasten”)





















