Download
1 / 15
170 likes | 1.04k Views
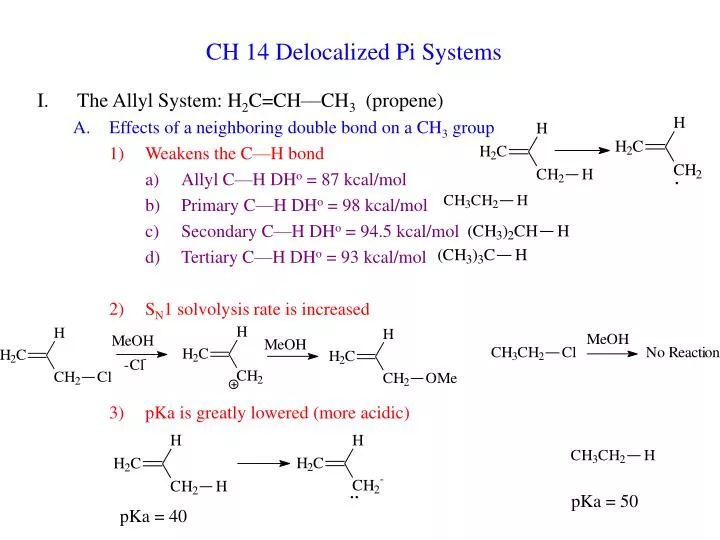
CH 14 Delocalized Pi Systems. The Allyl System: H 2 C=CH—CH 3 (propene) Effects of a neighboring double bond on a CH 3 group Weakens the C—H bond Allyl C—H DH o = 87 kcal/mol Primary C—H DH o = 98 kcal/mol Secondary C—H DH o = 94.5 kcal/mol Tertiary C—H DH o = 93 kcal/mol

E N D
CH 14 Delocalized Pi Systems • The Allyl System: H2C=CH—CH3 (propene) • Effects of a neighboring double bond on a CH3 group • Weakens the C—H bond • Allyl C—H DHo = 87 kcal/mol • Primary C—H DHo = 98 kcal/mol • Secondary C—H DHo = 94.5 kcal/mol • Tertiary C—H DHo = 93 kcal/mol • SN1 solvolysis rate is increased • pKa is greatly lowered (more acidic) pKa = 50 pKa = 40
Delocalization explains these observations • The three observations generate primary C., C+, and C- respectively. How can they be stabilized? • Delocalization = resonance structures spread the p-electrons over several C’s. This helps stabilize the radical, carbocation, and carbanion. • Molecular Orbital (MO) Representation of p-orbitals • H2C=CH—CH2 has each C sp2 hybridized • One unhybridized p-orbital for each carbon • C—C bonds are equivalent • Treat s-bonds as in Lewis structures
MO theory needed to describe the delocalized p-bond • 3 p atomic orbitals must give us 3 MO’s • We get a bonding (O nodes), a nonbonding (1 node), and an antibonding (2 nodes) MO • Fill up these orbitals with the appropriate number of p-electrons cation has 2 e-, radical has 3 e-, anion has 4 e- (B.O. = 1 for all)
4) Resonance Indicates that the charge or radical is found only on terminal C’s • MO theory shows differences only in pnb e- • That orbital has a node at C2 • MO theory agrees with the resonance hybrids • Allylic Reactions • Radical Allylic Halogenation • We would expect normal chlorination of propene (and we get it) • But, at low Cl2 concentrations, a radical reaction occurs for allylic groups
NBS Radical Bromination is the most common way to do this reaction • NBS = N-Bromosuccinimide • NBS gives a very low concentration of Br2 in CCl4 + HBr • Light (hv) or a radical initiator (RO.) needed to start the reaction • Allyl Radical resonance forms make two products possible. • Symmetric allyl groups give only one product • Assymetric allyl groups give product mixtures
Nucleophilic Substitution of Allylic Halides • SN1 is possible due to resonance stability of carbocation • At room temperature, major product is the less stable alkene • Kinetic Control = less stable isomer is formed faster through a lower energy transition state • Thermodynamic Control = most stable isomer is formed due to overall lower energy of products • Even though the less stable isomer is formed first under room temperature conditions (kinetic control), we can get the most stable product by heating the reaction for long periods (thermodynamic control). • H+ conditions mean the reaction is reversible • Allylic Alcohol reverts back to carbocation • Eventually, we will form the most stable product
f) Unstable isomer has lower Ea because its transition state is the most stable carbocation
SN2 Reactions of allylic halides are faster than for alkyl halides • Overlap of the p-bond with the p-orbital in the transition state stabilizes the T.S. for allylic reactants • Example:
Allylic Organometallic Reagents • Synthesis of Allylic Organometallic Reagents • Reaction with an Alkylithium reagent • Alkyl anion is more basic (less acidic) than allylic group • Alkyl anion deprotonates allylic group • TMEDA (like HMPA) is a good polar aprotic solvent • Normal Grignard Formation • Allylic Organometallic Reagents work as good 3C nucleophiles
Conjugated Dienes • Naming Dienes (compounds with two double bonds) • Allenes have both double bonds on the same C. H2C=C=CH2 • Two p-bonds on the same C must be perpendicular to each other • The central C is sp hybridized • Nonconjugated • Conjugated = two double bonds separated by a C—C single bond • All C’s are sp2 hybridized with a single p-orbital left • The 4 p-orbitals can overlap H2C=CH—CH=CH2
Naming is similar to alkenes, except diene at the end • Stability of Dienes 1) Conjugated dienes are more stable than nonconjugated dienes: DHo DHo = -30 kcal/mol DHo = -60 kcal/mol DHo = -57 kcal/mol
2) Conjugated Dienes are stabilized by resonance 3) Resonance Energy = 3.5 kcal/mol
Overlap of p-bonds Creates Conjugation • Four p-orbitals overlap over the entire molecule • Rotational barrier around the single bond is raised. Planarity is needed for conjugation. • The s-cis molecule has its p-bonds on the same side of C—C • The s-trans molecule has its p-bonds on the opposite sides of C—C • The “s” tells us there is a single bond between the C=C’s • The s-trans molecule is 3 kcal/mol more stable due to steric interactions in the s-cis molecule
The M.O. Picture for butadiene • 4 p-orbitals give 4 MO’s • Number of nodes increases up • 4 p electrons fill p1 and p2 • p-Bond Order = 2
Electrophilic Attack on Conjugated Dienes • Conjugated dienes are more reactive that alkenes • 3-chloro-1-butene is the normal Markovnikov product (1,2 addition) • 1-chloro-2-butene is the result of 1,4 Addition to a butadiene 2) It is common for dienes to give both 1,2 and 1,4 addition products