Download

1 / 19
190 likes | 340 Views
Structure Alignment in Polynomial Time. Rachel Kolodny Stanford University Nati Linial The Hebrew University of Jerusalem. Problem Statement. 2 structures in R 3 A={a 1 ,a 2 ,…,a n }, B={b 1 ,b 2 ,…,b m }

E N D
Structure Alignment in Polynomial Time Rachel Kolodny Stanford University Nati Linial The Hebrew University of Jerusalem
Problem Statement • 2 structures in R3A={a1,a2,…,an}, B={b1,b2,…,bm} • Find subsequences sa and sb s.t the substructures{asa(1),asa(2),…, asa(l)},{bsb(1),bsb(2),…, bsb(l)} are similar
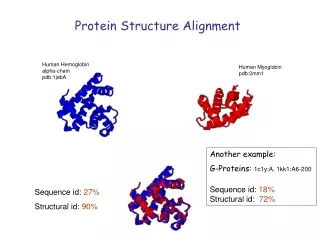
Motivation • Structure is better conserved than amino acid sequence • Structure similarity can give hints to common functionality/origin • Allows automatic classification of protein structure
Correspondence Position • Given a correspondence the rotation and translation that minimize the cRMS distance can be calculated Kabsch, W. (1978).
Position Correspondence • Given a rotation and translation one can calculate the alignment that optimizes a (separable) score • Using dynamic programming • Essentially similar to sequence alignment • Example score
Score cRMS • We want to give “bonus points” for longer correspondences • e.g. corresponding ONE atom from each structure has 0 cRMS • Even better scores ? • vary gap penalty depending on position in structure • Incorporate sequence information
Score cRMS A specific correspondence
Previous Work *most data taken from Orengo 94
“…It can be proved that, for these reasons, finding an optimal structural alignment between two protein structures is an NP hard problem and thus there are no fast structural alignment algorithms that are guaranteed to be optimal within any given similarity measure…” Adam Godzik ‘The structural alignment between two proteins: Is there a unique answer’ 1996 “There is no exact solution to the protein structure alignment problem, only the best solution for the heuristics used in the calculation.” Shindyalov & Bourne ‘Protein Structure Alignment by Incremental Combinatorial (CE) of the Optimal Path’ 1998
Exponentially many Focus on Scoring Functions
Exponentially many Focus on Scoring Functions
Exponentially many All Maxima are interesting Noisy data !!
Good scoring functions • Each of the functions is well-behaved • Satisfies Lipschitz condition • Thus, the maximum over a finite set is well-behaved • In each dimension two points at distance have function values that vary by O(n) • Need O(n) samples in every dimension
Polynomial Algorithm • Sample in rotation and translation space • compute best score (and alignment) for each sample point • Return maximum score • Need O(n6n2) time and O(n2) space
Internal Distance Matrices • Invariant to position and rotation of structures can be compared directly • Find largest common sub-matrices (LCM) whose distances are roughly the same
LCM is NP-complete • Harder than MAX-CLIQUE • Matrices encode distances that are positive, symmetric and obey triangle inequality
1dme 28 amino acids 1jjd 51 amino acids Example Best STRUCTAL score 149 Best score found by exhaustive search 197
Heuristic • Consider only translations that positions an atom from protein A on an atom of protein B • O(m*n) instead of O((n+m)3)