Download

1 / 45
450 likes | 643 Views
Tuberous Sclerosis Complex (TSC). Sergei Kashirny , MD LSU Neurology February, 3, 2011. Case.

E N D
Tuberous Sclerosis Complex (TSC) Sergei Kashirny, MD LSU Neurology February, 3, 2011
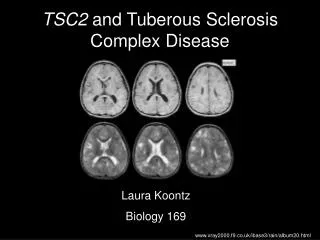
Case. 27 yo Asian female well-known in the clinic has been followed by neurology since infancy when she experienced infantile spasms. That condition was successfully treated with steriods and the patient remained seizure-free until age 10. At that age she had her first witnessed generalized tonic-clonic seizure. Her seizure became more frequent in the next 3 years and consist with CPS (complex partial seizure) and CPS with secondary generalization.
Except the presence of seizures she was the well developed child and had the average grades at school. • The patient had extensive work-up for her seizure including genetic analysis which led to the diagnosis of Tuberous sclerosis. • Valproate use led to control of the patient seizures up to the age 20 when she started to experienced them again. Currently she has about 1-2 seizures per year.
Family history: • maternal uncle has seizure since young age (unclear etiology) • 29 yo brother with cerebral palsy • Social : education – high school graduate, occassionaluse of ETOH, denies smoking or drugs.
Physical exam: • 3hypopegmented maculae on the back (about 3 x 5 cm) • No cardiac, pulmonary or renal abnormalities • Neurological exam is benign
Pathophysiology Clinically, TSC exhibits an autosomal dominant inheritance pattern, with a high spontaneous mutation rate. Two distinct genetic loci responsible for TSC have been identified: one on chromosome band 9q34 (also referred to as TSC1) and another on chromosome band 16p13 (TSC2). The TSC2 gene was identified in 1993, and its protein product has been named tuberin. Tuberin has GTPase-activating properties and seems to function as a tumor suppressor. The highest levels of tuberin are found in adult human brain, heart, and kidney. Individual tubers are thought to arise developmentally when mutated neural progenitor cells in the subependymal germinal matrix give rise to abnormally migrating daughter cells that in turn produce tubers. The tubers may undergo cystic degeneration or calcification, or exhibit contrast enhancement on neuroimaging, but these features do not necessarily imply malignant transformation.
Hamartin, the TSC1 product, was identified in 1997 and may also function as a tumor suppressor. Hamartin and tuberin together form a tumor suppressor complex, which, through the GTPase activating function of tuberin. Subsequent mutational analysis has shown TSC2 mutations to be present in 80-90% of affected individuals, while TSC1 mutations are present in 10-20%. It is found a higher incidence of "mental handicap" in persons with TSC2 mutations than in those with TSC1 mutations. A TSC2 genetic abnormality was found to be associated consistently with more severe clinical disease regardless of organ system. Although prominent phenotypic variability was still the rule, patients with TSC2 abnormalities were more apt to have higher tuber counts, refractory seizures, autism, larger cardiac rhabdomyomata, and more severe cutaneous lesions. This suggests that, while tuberin and hamartin have similar functions, tuberin plays a more critical role in regulation of cellular differentiation.
The high incidence of sporadic TSC, coupled with a probable "second hit" phenomenon, seems a likely explanation for the marked phenotypic variability observed. The second hit hypothesis suggests that in addition to an inherited or sporadic autosomal mutation in one allele of either TSC 1 or TSC 2, clinical signs and/or symptoms manifest only after a further mutation or inactivating event in the second, unaffected allele (“second hit”). This allows considerable potential for diversity, not only among various deletions and mutations between 2 genetic loci, but also with regard to possible interactions between protein products of varying functionality arising from different mutations on each allele. Further complicating the high spontaneous mutation rate is the observation that parents of an affected child, who themselves show no sign of TSC, nonetheless have an increased risk (approximately 2% overall) of having additional affected children.
Frequency Prevalence - 1 case per 10,000 population. Race TSC affects all races without a clear-cut predominance. Sex TSC affects both sexes equally. Age TSC can present at any age.
CLINICAL PRESENTATION • In 1908 Vogt set the triad of intractable epilepsy, mental retardation, and adenoma sebaceum; this description (until relatively recently) represented the hallmark of tuberous sclerosis complex (TSC) to most clinicians. • As many as 50% of people with TSC have normal intelligence, and increasingly the diagnosis is being newly made in adults with renal, cutaneous, or pulmonary manifestations.
Clinical Manifestations of TSC • Brain: cortical tubers, subependymal nodules, subependymal giant cell astrocytomas • Eye: retinal hamartomas • Heart: cardiac rhabdomyomas • Kidney: benign angiomyolipomas, cysts, malignant angiomyolipomas, renal cell carcinoma • Lung: lymphangioleiomyomatosis, mutifocalmicronodularpneumocyte hyperplasia • Skin: hypomelanoticmacules, shagreen patches, periungual or subungualfibromas, facial angiofibromas • Behavior: mental retardation, autism, bipolar disorder
Diagnostic criteria • Definite TSC - Either two major features or one major feature plus two minor features • Probable TSC - One major plus one minor feature • Possible TSC - Either one major feature or two or more minor features
PHYSICAL:Neurological findings • Seizures (90%) • Mental retardation and learning difficulties (50-60%) • Sleep disorders (60%) • Autism and behavioral difficulties (30-50%) • Subependymal giant cell astrocytoma (15-20%)
TSC frequently presents with infantile spasm (generalized epilepsy occurring in infants). • Infantile spasms are significantly associated with long-term developmental delays and mental retardation. • Poor seizure control or ineffective control of the infantile spasms was associated with the highest risk for poor developmental outcome.
Epilepsy in TSC • about 90% of patients will experience seizures • About 20%-30% of those patients have medically intractable epilepsy • Aggressive treatment is necessary to reduce risk for negative neurological outcome (development, cognition, behavior)
Skin findings • TSC is considered a neurocutaneous disorder, and another hallmark of this disease is facial angiofibroma. This used to be called adenoma sebaceum, but it has nothing to do with an adenoma or with excessive sebum or acne. These are angiofibromas -- cutaneousfibromas -- that typically occur in both cheeks in a butterfly distribution. These lesions can become quite large. In some patients, they can result in bleeding and significant cosmetic impairment.
Cardiac findings: • Cardiac involvement is usually maximal at birth or early in life; it may be the presenting sign of TSC, particularly in early infancy. 50-60% of individuals with TSC have evidence of cardiac disease, mostly rhabdomyomas. • Rhabdomyomas are benign tumors that may be focal or diffuse and infiltrating in character. They produce symptoms primarily through outflow tract obstruction or by interfering with valvular function • Rhabdomyomas develop during intrauterine life (usually between weeks 22 and 26 of gestation) and can result in nonimmunehydropsfetalis and fetal death. The majority of cases, however, are clinically asymptomatic. • The lesions typically undergo spontaneous regression in the first few years of life, although residual areas of histologically abnormal myocardium may persist.
Ocular findings: • Up to 50% of patients have ocular abnormalities. These lesions are retinal astrocytomas that tend to become calcified over time. They appear as rounded, nodular, or lobulated areas on funduscopic examination, becoming whitish in color as they calcify.
Pulmonary findings: • Symptomatic pulmonary involvement occurs almost exclusively in adult women, generally aged 30 or older. Recent studies have found cystic pulmonary abnormalities in 40% of women with TSC. Three forms have been described: multifocal micronodularpneumocyte hyperplasia, pulmonary cysts, and lymphangiomyomatosis.
Renal findings: Renal manifestations are the 2nd most common clinical feature. • Polycystic kidney disease • in 2-3% of persons with TSC • presents early in life with hypertension, hematuria, or renal failure • occurs as the result of a genetic abnormality affecting both the TSC2 gene and the PKD1 gene adjacent to it • have relatively little functional renal tissue, and ultimately require renal transplantation • highly susceptible to complications of UTI or nephrolithiasis. • Renal cysts (as opposed to polycystic kidney disease) are found in 20% of males and 10% of females with TSC. They are rarely if ever symptomatic. • AMLs in 80% of persons with TSC. They also can occur as isolated lesions in persons without TSC. • Renal cell carcinoma
Other organ systems • Gingival fibromas - • 70% of adults • 50% of children with mixed dentition • Hepatic cysts and AMLs • asymptomatic and nonprogressive, • 24% of patients • marked female predominance • Arterial aneurysms • small number of patients • Intracranially, in the aorta and axillary arteries
Work-up • CT/MRI brain • every 2 years in asymptomatic patients, at least until puberty • Renal ultrasound • To assess AMLs or cysts • Due to under-recognition and underestimation of AML occurrence and size, renal ultrasound is losing favor and is being replaced by abdominal MRI. • abdominal MRI - every 2-3 years • Echocardiogram • Baseline evaluation • Usually not repeated if no lesions • EKG • Baseline evaluation and then every 2-3 years
EEG • Evaluate for seizure • Repeat as clinically indicated • PET scan • Evaluation for epilepsy surgery • Scanning with alpha-methyltryptophan to identify epileptogenictubers • Molecular genetic testing • Commercially available • Under optimal circumstances, identifies mutations in 75-80% • Negative genetic test result does not exclude a diagnosis • Diagnosis should be made in most cases using established criteria • Genetic testing is useful in uncertain or questionable cases: • for prenatal diagnosis • for screening family members of an affected individual • The utility of molecular diagnostic testing is limited by the cost
MEDICAL TREATMENT • Serolimus (rapamycin) • Everolimus – FDA has granted accelerated approval (11/2010) for SEGA that cannot be treated with surgery
Serolimus (Rapamycin): • almost 50% decrease in renal angiomyolipomas (AML) volumes by the end of the 12-month rapamycin administration period • improvements in forced expiratory volume (FEV1), forced vital capacity (FVC) and residual volume (RV) in patients with pulmonary lymphangioleiomyomatosis (LAM). • Described regression of subependymal giant cell astrocytomas (SEGA) in association with rapamycin. • Everolimus: • reduced SEGA volume by 30% in 78% patients; and by at least 50% in 32% patients in the first 6 months of treatment • reduced SEGA volume by about 50% by 18 months • improvements in seizure control: nearly 20% of patients experiencing seizure freedom and more than 50% experiencing a more than 50% reduction in seizure frequency • improvement in quality of life measures and no change in neuropsychiatric parameters.
Infantile spasns in TSC • Vigabatrin is the treatment of choice in TSC • Irreversible inhibitor of GABA transaminase • Risk for adverse effect on peripheral vision • As high as 95% response rate in TSC • Steroids (ACTH or oral prednisone) • Valproate, topiramate, clonazepam minimally effective as single agents but may have beneficial adjunctive use
Epilepsy treatment • The main complication of TSC requiring long-term therapy is epilepsy. • Antiepileptic medications (AEDs) are the mainstay of therapy for patients with TSC. • No one medical treatment gives satisfactory relief for all patients. • A combination of medical treatment modalities frequently is required. • The choice of specific AED(s) for treating seizures in patients with TSC is based on the patient's seizure type(s), epilepsy syndrome(s), other involved organ systems, age of the patient, and side effect profiles and formulations available. • Vigabatrinis the drug of choice for children with TSC and infantile spasms. • Long-term use of benzodiazepines or barbiturates should be avoided. These drugs often aggravate underlying behavioral or cognitive problems and have many less toxic and often more effective alternatives. • Carbamazepine, oxcarbazepine, and phenytoin may cause exacerbation of seizures, particularly in younger children and infants, and can precipitate or aggravate infantile spasms. They should not be used in children with TSC who are experiencing infantile spasms.
Epilepsy surgery • Aggressive treatment should include the epilepsy surgery: • 60% of patients can expect to be seizure-free • 10% would have rare seizures • 10% would have a greater than 90% reduction
SURGICAL TREATMENT • Focal cortical resection • Corpus callosotomy • Can be effective in reducing atonic and tonic seizures (ie, drop attacks) • typically is not helpful for other seizure types • Seizure freedom following corpus callosotomy is rare but can occur. • Vagus nerve stimulation • Simple and complex partial seizures appear to respond better than partial seizures with secondary generalization. • SEGA resection • When produce hydrocephalus or significant mass effect • If a gross total resection can be achieved, recurrence is unlikely