Download

1 / 61
660 likes | 830 Views
A Hybrid Approach for the Automated Finishing of Bacterial Genomes.

E N D
A Hybrid Approach for the Automated Finishing of Bacterial Genomes Ali Bashir, Aaron A Klammer, William P Robins, Chen-Shan Chin, Dale Webster, Ellen Paxinos, David Hsu, Meredith Ashby, Susana Wang, Paul Peluso, Robert Sebra, Jon Sorenson, James Bullard, Jackie Yen, Marie Valdovino, Emilia Mollova, KhaiLuong, Steven Lin, Brianna LaMay, AmrutaJoshi, Lori Rowe, Michael Frace, Cheryl L Tarr, Maryann Turnsek, Brigid M Davis, Andrew Kasarskis, John J Mekalanos, Matthew K Waldor& Eric E Schadt Presented by George Roberts III
Infectious Disease: A Complex Phenomenon Ecology Microbial Genomics Pathobiology
Seven Pandemics • References in antiquity • Hippocrates • Galen of Pergamon • Local disease • Seven pandemics since 1817 • Tens of millions of deaths • 1 – 6 originated and 7th incubated in subcontinent • “classic” biotype (1817-1923) (nonhemolytic O1) • CTXΦclass on small chromosome and CTXclassΦ-CTXclassΦ on large chromosome • Defective due to structural genomic issues (can’t initiate rolling circle replication) • El Tor: seventh pandemic (1961-1975…) • First isolated in 1905 from six Hajji – Jabal al Tor (Sinai) • Lead and corresponding authors are from Mt.Sinai NYC • Highly-infective, low mortality (Sulawesi 1938) • Hemolytic O1 • SXT family of antibiotic resistance elements • > 570,000 cases worldwide
El-Tor Strains • N16961 • Isolated in 1971 in Bangladesh • “cannonical” reference genome • Possesses GI-12, GI-14, GI-15 and κ-phage island • CIRS101 (Dhaka, 2002 – ACVW00000000) • Sequenced strain most closely related to H1 • Displaced other clones – reasons unknown • CtxB of classical origin • Missing GI-12, GI-14, GI-15 and κ-phage island • Greater infectivity than closely related strains (Colwell group 2010) • H1 – 2010 Haitian outbreak • Resembles Asian strains of the last decade • Hundereds of fatalities • O139 (Non - El Tor): first non-O1 epidemic cholera – not pandemic cholera • SXT (self-transmissible conjugative)-related Integrating Conjugative Element (ICE) • Horizontal gene transfers with El Tor
Nepalese Origin of H1 • Rumors that cholera was brought by UN Peacekeepers of Nepalese origin • sparked riots • Relief efforts disrupted • Group V is monophyletic Hendriksen et al. 2011 mBio2:e00157-11 = Bangladesh
Microbiological Theme • Evolution is discontinuous • Horizontal gene transfer is a game changer • Pandemics • genetic • social • migratory Image: PrzykutaCreative Commons Attribution/Share-Alike License
Vibrio cholerae • Vibrio – genus of curved Gram negative rods • Vibrare: [L] to vibrate • facultative anaerobe • Spread by poor sanitation / seafood • Rehydration therapy / antibiotics • Two circular “chromosomes” • Several horizontal gene transfer events Image credit: popular logistics
Cholera Toxin (Ctx) is CTXφ-encoded prophage lysogeny image credit: Wikipedia commons – Suly12 Filamentous phage image credit: Tikunova and Morozova, ActaNature
Ctx Enzymatic subunit - A1 Cl- Cl- GM1-binding subunits - B5 H20, K+, Na+ and HCO3- Cl- Cl- Cl- Cl- Cl- Intestinal lumen GM1 ganglioside Cl- CFTR Cl- Cytoplasm Cl- [↑ 100x cAMP] A1+ARF6 endocytosis PDI A1-Arf6 A1B5 A1 + A2B5 AC ADP ribosyl-AC Ctx crystal structure: Zhang et al. 1995 JMB 251:563-73 Pathway artwork: GGRIII
Toxin-coregulatedPilus (TCP) • Encoded by the VPI • Expressed with CTX • Required for CTXφ infection
Satellite Phage • Other phage provide required factors • Toxin-like Cryptic (TLC) region • TLC-Knφ – a filamentous satellite phage of fs2φ • Integrates into dif-like site • Can restore dif-V. cholerae to dif+ CTXφ-susceptible • RS1φ • Related to CTXφ • Overlap with CTX in classical strains prevents CTX replication
Sanger vs. Next-generation Sequencing • Sanger dideoxy (1977) • High accuracy • 500-700bp reads • sequencing by termination • Increases amount of template required • Limits read length • Next-generation sequencing (1999) • Sequencing by synthesis • Ultra high-throughput • Very short (40bp), med. (~300bp) or very long reads (23kb)
Illumina • Short reads ~ 40 bp • Highest fidelity (99.5%) • Competition: all four dNTPs are present • “Lawn” of adapter oligos • Detects methylation of sulfite-treated DNA • Detects protein-binding • Reads a single base each cycle (rev. terminator) • High-performance for runs of a single nucleotide Metzker (2010) Nature Reviews Genetics 11, 31-46
Illumina • DNA is sheared • ends repaired • 3’-A overhang • Adapters ligated
Illumina 3’ ends are extended Exponential “bridge” amplification creates myriad localized “rainbow” structures
Illumina Competitive addition of n+1 dNTP-fluor Wash unincorporated dNTPs Cleave fluor / read fluoresence
454 group • Pyrosequencing • dNTPs added sequentially • Pyrophosphate release is detected by luciferase • Medium reads ~329 bp • Moderate fidelity (98.7%) • Poor performance for runs of a single nucleotide • Bead-based
--------------iterative addition--------- --dATP dTTP dGTP dCTP -- Pyrosequencing (454) Adenosine 5’-phosphosulfate + Image credit EMBL:EBI
454 group Shear DNA Ligate A/B capture fragments on beads adapters
454 group linear!
Pacific Biosciences • Phospho-linked fluorophores • Cleaved during incorporation • Higher speed, fidelity and processivity • LiCor, Life/VisiGen • Long reads, up to 23kb (mean of 2-6 kb) • Low-fidelity (~84%) • Exceptionally useful for assembly • SMRT - “Eavesdropping on the polymerase” Metzker (2010) Nature Reviews Genetics 11, 31-46
Pacific Biosciences Polymerase • φ29 polymerase • processive - >70 kb • Stable, single subunit, high-speed • Efficient with phospholinkeddNTPs • Minimal context bias in WGA by strand displacement • Sequencing processivity • Laser damage (strobe reads) • Altered substrate • Immobilization
Pacific Biosciences Chemistry Φ29 pol
Pacific Biosciences ZMW • Zero-mode Waveguide (ZMW) • Confines excitation to 20 zeptoliters (20x10-21) • Enables optimal [substrate] • Fluoresecence detected during incorporation (msec) • Diffusion rapidly dissipates signal / ready for next base
Some Assembly Required… http://www.icrisat.org/ceg/bt-workshops/dedwads-genomic-resources.pdf
Alignment methods AGATCCGATGAG • De Bruijn graph • Developed for SBH • Excellent for short reads, high-coverage & high accuracy • Overlap-layout-consensus • Longer reads (Sanger & 3rd generation) • Lower coverage • Lower accuracy • Combine scaffolding, overlap and error-correction • Long reads aid assembly of short, high accuracy reads AGAG GAGGCTTTAGA AAGTCGAG GAGACAA ..ACGATTACAATAGGTT.. Image credits: HamidrezaChitsaz
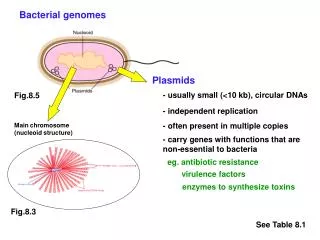
State of Sequenced Genomes • 26% of bacterial genomes are “complete” • Large-scale structural and linear organization? • Small genomic differences have major effects… • Repetitive regions: CTX prophage
PacBio Reads • Standard • R&D version of the PacBio DNA Sequencing 1.0 kit • 75 to 120 minutes • C2 Chemistry • Replaced by RS
PacBioReads • Paired Reads Schematic of a SMRTbell™ template. Travers K J et al. Nucl. Acids Res. 2010;38:e159-e159
PacBio Reads • Strobe Reads • Decrease damage to φ29 pol • Lower throughput R&D instrument • Two dark periods • three sequence islands • Distance in dark period is estimated • Read times • 4-48-4-48-8 • 4-52-4-52-4 • Becoming “obsolete” • Abasic sites in SMRTbell hairpins to prevent pol “wrapping” • RS chemistry: avg. 2700bp & 5% of reads >5100 bp
2010EL-1786 from Haiti (CDC) Accession contigscoverageN50 • AELH00000000.1: 107 98.84% 151kb • AELJ00000000.1: 105 98.96% 154kb • AELI00000000.1: 93 98.94% 155kb • 99.99% identity
Repetitive Region: rRNA operons • Seven 5kb rRNA operons (98.04%-99.94% identity) • Account for 7 of 45 gaps in Illumina/454 sequence • Each was spanned by >3 strobe reads • Multiple overlapping C2 reads
Finishing Sanger / PCR fill in • 78 gaps • 56 gaps < 600 bp (easily within Sanger range) • 55 Successful PCR confirming correct contig order • 48 had no non-specific products (casualties of high-throughput) 48 x FR = 96 • Sanger sequencing used to fill in gaps • 1 of 48 did not produce sequence
Superintegron • Encodes a phage-related [Y]-recombinase – intI • attI x attC • Discovered in V. cholerae in 1998 • Reapeat-rich – comparative structure difficult • ORFs of bacterial, viral or eukaryotic origin • Passed among Vibrio genus… • Antibiotic resistance, toxins • Highly variable http://www.wwnorton.com
Superintegron • CDC contigs are fragmented (repetitive seq.)
Integrating Conjugative Element (ICE) • Prevalent in Asian V. cholerae since the emergence of O139 in 1992 • Recent O139 ICE elements lack antibiotic resistance
Repetitive Region: CTX, RS1 & TLC • RS1 element • Sequence similarity to CTXφ • Often interspersed with CTXφ • CTX prophage • H1 & CIRS101: RS1 upstream of CTX – can’t replicate • Transposase adjacent to CTX • Characteristic of recent seventh pandemic isolates with classical ctxB (CIRS101, H1 and its hypothesized progenitor) • Tandem TLC • Confirmed by Southern blotting
H1 Genome Summary • A patchwork of elements with various origins • Multiple lysogenic events • CTXΦ • TLCΦ • Mobile elements • Super integron • Type VI secretion system (T6SS) • Similar to phage transcellular injection mechanism • 10-5 – fold reduction in E. coli
Assembly • Generated a consensus CDC contig set (control) • Assembled de novo in parallel
Error Correction • Contig edge correction using short sequences • Subgraph untangling • Assemble contigs into scaffolds • Repeat untangling and assembly
SubgraphUntagling • Supplementary Figure 13. Examples of subgraph untangling. The first column shows the graph before a particular untangling operation, the second after that operation. A) The scaffold link between contigs S and K contain the smaller internal contig I. This spanning link can be eliminated leading to a simple linear path. B) Multiple contigs exist between S and K. Since all internal contigs (I1 to Im) are connected to both S and K we can order them in a direct path from S to K based on their layout. C) A repeat contig R is resolved with a scaffolding edge between S and K. Contig R is duplicated and its remaining edges are removed from the original contig R and passed onto the duplicated node. D) A link between S and K exists but the internal nodes are not completely connected to either S or K (or both). In this case edges are inferred between the source and sink nodes, and all internal nodes, based on the span distributions of linking edges and the lengths of the internal nodes.