Download

1 / 46
460 likes | 470 Views
An Introduction to X-Ray Diffraction by Single Crystals and Powders Patrick McArdle NUI, Galway, Ireland. The Nature of Crystalline Materials. Crystalline materials differ from amorphous materials in that they have long range order. They also exhibit X-ray powder diffraction patterns .

E N D
An Introduction to X-Ray Diffraction by Single Crystals and Powders Patrick McArdle NUI, Galway, Ireland pma 2019
The Nature of Crystalline Materials • Crystalline materials differ from amorphous materials in that they have long range order. They also exhibit X-ray powder diffraction patterns. • Amorphous materials may have very short range order (e.g. molecular dimers) but do not have long range order and do not exhibit X-ray powder diffraction patterns. • The packing of atoms, molecules or ions within a crystal occurs in a symmetrical manner and furthermore this symmetrical arrangement is repetitive throughout a piece of crystalline material. • This repetitive arrangement forms a crystal lattice. A crystal lattice can be constructed as follows: pma 2019
A 2-dimensional Lattice Pick any position within the 2 dimensional lattice in Fig. 1(a) and note the arrangement about this point. Place a dot at this position and then place dots at all other identical positions as in Fig. 1(b). Join these lattice points using lines to give a lattice grid. The basic building block of this lattice (unit cell) is indicated in Fig. 1(c). pma 2019
c b a Orthorhombic Unit Cell Types and The Seven Crystal Systems Cubic a = b = c. = = = 90º. Tetragonal a = b c. = = = 90º. Orthorhombic a b c. = = = 90 º. Monoclinic a b c. = = 90º, 90º. Triclinic a b c.. 90º. Rhombohedral a = b = c. = = 90 º. (or Trigonal) Hexagonal a = b c. = = 90º, = 120º. In general, six parameters are required to define the shape and size of a unit cell, these being three cell edge lengths (conventionally, defined as a, b, and c), and three angles (conventionally, defined as , , and ). In the strict mathematical sense, a, b, and c are vectors since they specify both length and direction. • is the angle between b and c, is the angle between a and c, is the angle between a and b. The unit cell should be right handed. Check the cell above with your right hand Unit Cells may centred on one or more faces or at the centre of the cell. When the unit cells listed above are combined with centring there are 14 different Bravais lattices. pma 2019
Cubic Tetragonal Orthorhombic Body Centred Cell Face Centred Cell Monoclinic I F End Face Centred Cell Triclinic C Trigonal Hexagonal Primitive Cell P The 14 Bravais Lattices pma 2019
Fig. 2 Fig. 3 Primitive and Centered cells On the previous slide you can see that that a Monoclinic unit cell can be primitive or centred (by convention on the c face). These are referred to as Monoclinic P or Monoclinic C. Choice of Unit Cell A unit cell can be any unit of a lattice array which when repeated in all directions, and always maintaining the same orientation in space, generates the lattice array. There is no unique way of choosing a unit cell. For example, each of the cells (A to D) in Fig. 2 are OK. However, the cell favoured by crystallographers is smallest most orthogonal cell that displays all of the symmetry of the lattice. Thus, cells C and A are the preferred unit cells for the lattices of Figs. 2 and 3 respectively. pma 2019
Four important points on crystal lattices: 1. Every crystal system has a primitive Bravais lattice or P type. 2. The distribution of lattice points in a cell must be such as to maintain the total symmetry of the crystal system. Thus, the cubic system cannot have a C-type cell. 3. The fact that a unit cell meets the symmetry requirements of a crystal system does not guarantee its inclusion within the crystal system. This could result if the lattice it generated could be equally well represented by a unit cell type which is already included within the crystal system. The C-type cell for the tetragonal system (see Fig. 4) provides a good example. 4. If you apply point 3 to the orthorhombic system you will find that the primitive cell you generate will not have 90º angles. This would not be orthorhombic and thus orthorhombic C is included in the orthorhombic system. This is shown on the next slide. pma 2019
Tetragonal C and Orthorhombic C centred cells A simplified view down c-axis can be used to illustrate points 3 and 4 on the previous slide. Orthorhombic a ≠ b ≠ c, a = b = = 90º Tetragonal a = b ≠ c, a = b = = 90º a a b b Smaller cell is Tetragonal P and is preferred Angle not 90° thus the smaller cell is not orthorhombic and must be rejected pma 2019
Symmetry - Point Groups and Space Groups z • Point Groups describe symmetry about a point. • Formaldehyde has the symmetry of the C2v point group. • The symmetry operations of C2v are C2 (rotation about z of 360/2), two planes of symmetry sv and sv’ (vertical planes) and the identity operation. • C2v is the Schoenflies symbol for the point group • mm2 is the Hermann-Mauguin symbol for this point group. • Stereographic projections can be used to represent point groups • There are 32 crystallographic point groups (also called classes) • Point groups cannot describe a crystal lattice – Space Groups are required. y C2 O x C H H sv’ sv + + + + Stereographic projection of C2v or mm2 (pick any + and apply C2 and s to get the others) pma 2019
The reduction of a space group to a point group is described on slide 16 pma 2019
Space Groups from Point Groups. • Point Group symmetry operations (sym.ops.) • Identity x,y,z → x,y,z • Inversion x,y,z → -x,-y,-z • Mirror e.g. xy plane mirror x,y,z → x,y,-z • Rotation axis rotation by 360/n n = 1,2,3,4 or 6. • Space Group sym.ops. also have translational symmetry – screw axes and glide planes • A screw axis is represented by nm where n is the rotation (360/n) and m/n is the fraction of the unit cell length of the translation e.g. a 21 along b • 21 along b x,y,z → -x,1/2+y, -z • A glide plane has translation (often ½) and a reflection • b glide with a yz mirror x,y,z → -x,½+y,z • Combining these symmetry operations with the 32 point groups leads to the 230 possible 3d Space Groups. pma 2019
CRYSTAL SYSTEMS (7) BRAVAIS LATTICES (14) SPACE GROUPS (230) P 15 36 Cubic F 11 I 10 P 49 68 Tetragonal I 19 P 30 F 5 59 Orthorhombic I 9 C and A 15 P 8 13 Monoclinic C 5 2 Triclinic P 2 25 Rhombohedral P and R 25 27 Hexagonal P 27 Fig. 9 The 230 Space Groups The distribution of Space Groups among the Bravais lattice type is shown in Fig. 9. The International Tables for Crystallography list the symmetry properties for all 230 Space Groups. The 2nd edition was in one volume and edited by Kathleen Lonsdale. The current edition runs to 7 volumes. The CSD or Cambridge Data Base is a repository for the structures of organic and organometallic compounds which in 2019 exceeded 1000000 entries. Space Group determination is an important step in crystal structure determination. pma 2019
The ABSEN program within Oscail can provide Bar Charts of the contents of the Cambridge Data Base (CSD) No 14 most entries The number of entries by crystal system Entries in the first 25 space groups pma 2019
Space Group No. 14 P21/c • This monoclinic space group has the most entries on the CSD • Read its name as “p21 upon c” • The full name is P 1 21/c 1 (there is no symmetry on a or c) • There are 4 general positions 1 x,y,z 2 -x,1/2+y,1/2-z 3 -x,-y,-z 4 x,1/2-y,1/2+z (21 at 0,y,1/4) Inversion at (0,0,0) Glide at (x,1/4,z) 2 4 glide normal to screen at 1/4b 1 3 View down a Stereographic view down b 21 screw axis inversion centre glide plane at 1/4b pma 2019
Space Group No. 14 P21/c, Example Benzoic acid CSD BENZAC a 0 1 • The molecules in the unit cell of benzoic acid illustrate the positions in the stereographic projection of the space group • 1 and 2 are related by a 21 screw • 1 and 3 by an inversion centre • 1 and 4 by a c glide 2 1 2 4 4 3 3 c pma 2019
Converting a Space Group to a Point Group • When the translational parts of the symmetry operations are removed the Space Group is reduced to a point group • The P21/c symm ops are; 1, x,y,z 2, -x,½+y,½-z 3 -x,-y,-z 4 x,½-y,½+z • Removing the ½ s will remove the translations leaving • 1, x,y,z 2 -x,y,-z, 3 -x,-y,-z 4 x,-y,z • With respect to 1 symm ops 2,3 and 4 now are a 2-fold axis along b, inversion and a mirror normal to b. Thus the HM symbol for this is 2/m and the Schoenflies symbol is C2h • Thus P21/c is reduced to the point group 2/m. 2/m is the only centrosymmetric monoclinic point group Stereographic projection of 2/m pma 2019
Crystal Planes and Miller Indices The use of crystal planes to describe the structure of crystals goes back to the start of crystallography and crystal planes were used by Bragg to explain diffraction as will be seen later. Crystal planes are defined by the intercepts they make on the crystal axes of the unit cell. The inverse of these fractions are the Miller Indices of the planes. In (a) the intercepts are ½, ½, 1 and the Miller Indices are (2 2 1). In (c) the intercepts on b and c are at infinity the inverse of which is 0 and the plane is the (2 0 0). In (f) the plane cuts the negative c axis at -1 and thus is (1 1 -1). In crystallography -1 is often written ī and pronounced “Bar 1”. pma 2019
Diffraction and the Bragg Equation Max von Laue was the first to suggest that crystals might diffract X-rays and he also provided the first explanation for the diffraction observed. However, it is the explanation provided by Bragg that is simpler and more popular. In the Bragg view crystal planes act a mirrors. Constructive interference is observed when the path difference between the two reflected beams in (a) = nl. The path difference in (a) is 2my. Since my/d = sin 2my = 2dsin = nl where d is the interplanar spacing. pma 2019
Bragg Reflection Order On the previous slide in (a) it is clear that the planes are the (1,0,0) set of planes. If the path difference is simply one wavelength the Bragg condition can be stated as This is a first order reflection. If the path difference is two wave lengths the Bragg condition becomes and the reflection is a second order reflection. pma 2019
Step by Step Single Crystal Structure Solution using X-ray Diffraction • Bragg's equation specifies that, if a crystal is rotated within a monochromatic X-ray beam, such that every conceivable orientation of the crystal relative to the beam is achieved, each set of planes will have had the opportunity to satisfy the Bragg equation and to give rise to a reflection. • In order to solve a crystal structure it is necessary to record a large number of reflections. • Many experimental techniques have been devised to achieve this. The steps involved in a crystal structure determination are summarised in the flow chart on the right. • When you have had a look at the introduction to single crystal X-ray diffraction given here you can look the worked examples in Oscail tutorials on Crystallogtaphy. pma 2019
Single Crystal X-Ray Data Collection The first crystallographic data collection systems used photographic methods. Moderndiffractometers use electronic area detectors which measure hundreds of reflections at a time. In a typical setup a crystal is oscillated over < 2° while an image is collected the crystal is then rotated by the same amount and oscillated again. The process is repeated over a total range of up to 180 or 360°. Data collection time depends on the crystal size, quality and other factors and may be from 40 mins to several hours on a lab diffractometer and just seconds on a synchrotron. pma 2019
Determination of the Lattice type and Space Group High symmetry can lead to reflections being systematically absent from the data set. Absent reflections have no measurable intensity. There are two types of absences, General Absences and Special Absences. The general absences determine the lattice type; Primitive (P) has no general absences and no restrictions on h, k or l. End Cantered (C) h+k=2n+1 are all absent. Face Cantered (F) only h, k, l, all even or all odd are observed. Body Cantered (I) h+k+l=2n+1 are all absent. The special absences refer to specific sets of reflections and are used to detect the presence of glide planes and screw axes. Some Space Groups are uniquely determined by special absences but in many cases several Space Groups will have to be considered. Computer programs are able to lay out the data in tables with absences indicated and possible Space Groups can be suggested however the choice of Space Group will often need to be carefully considered. pma 2019
Reflection Analysis I/I Cut1 Cut2 Cut3 = 3.0 6.0 12.0 Group Cond. Op. All Odd Cut1 Cut2 Cut3 Op. No. h00 h=2n+1 21.. 18 10 8 8 8 1 0k0 k=2n+1 3 1 1 0 0 .21. 2 00l l=2n+1 11 6 0 0 0 ..21 3 0kl k=2n+1 b.. 95 53 49 44 37 4 0kl l=2n+1 c.. 49 43 40 33 5 0kl k+l=2n+1 n.. 40 34 30 28 6 h0l h=2n+1 .a. 412 211 96 89 81 7 h0l l=2n+1 211 1 1 0 .c. 8 h0l h+l=2n+1 .n. 212 95 88 81 9 hk0 h=2n+1 ..a 168 84 67 60 49 10 hk0 k=2n+1 ..b 84 76 69 48 11 hk0 h+k=2n+1 ..n 86 71 69 55 12 hkl k+l=2n+1 A.. 1591 1196 1084 902 13 hkl h+l=2n+1 .B. 1638 1271 1151 915 14 hkl h+k=2n+1 ..C 1651 1285 1145 921 15 hkl h+k+l=2n+1 I 1637 1288 1148 943 16 hkl not all odd/even F 2440 1876 1690 1369 17 P21/c (14) CSD Total for all SGs in 2019 has exceeded 1000000 In this case P21/c is the only choice offered and this is likely to be correct. Notice the symmetry operations move to the right when present in the data. pma 2019
h0l zero level reflections for a P21/c example 300 reflection pma 2019
Solving the Structure The unit cell, the Space Group and the reflection intensities cannot be used to generate the structure as there is no reflection phase information in the data set. This is the phase problem. If the reflection phases were known then an electron density map could be calculated using a Fourier series. If (x,y,z) is the electron density at x,y,z then The F here is the square root of the measured intensity When intensity is measured it is measured without sign and the phase is lost. • There are three ways to solve the phase problem: • The Patterson or heavy atom method • Direct Methods (Hauptman and Karle 1985 Nobel prize) • The Charge flipping method is a recent development The ShelxT program combines these methods automatically, solves the structure and suggests space groups. pma 2019
Data Resolution It is the amount of 4s data at 1Å that determines the success of direct methods this example has sufficient 4s data for direct methods. pma 2019
Refining a Structure It should be possible to “see” atoms in an electron density map if it has good resolution i.e. at least 1Å resolution. The steps in refining a structure are. 1. Use whatever atoms you have that look OK to generate an electron density map. 2. The known atoms are subtracted from this to generate a difference map. 3. Any atoms that have been missed should be in the difference map. 4. The refinement process minimises the difference between observed and calculated reflection intensities. 5. In the final difference map there should be no peaks larger than a H atom i.e. > 1e/Å3. (A H atom has a volume of about 1Å3 and has 1 e.) Resolution The resolution of a crystal structure is usually quoted in Angstroms, Å. Standard small molecule structures should always be at least of 1 Å resolution to give accurate bond lengths. Resolution can be related to Bragg angle at any wavelength through the Bragg equation n = 2d sin. Using the value of the reflection with the largest Bragg angle in a data set then d = l/2sin gives the resolution. The pattern shown on slide 15 has a resolution of 0.98Å at the edge. pma 2019
Anisotropic refinement of the non-hydrogen atoms – In the early stages atoms are refined as if they were spheres. Since atoms vibrate in a way that is controlled by chemical bonds and interactions with their neighbours, it is better to refine then as ellipsoids. One parameter (the radius) is enough to define a sphere this with x,y,z means that isotropic refinement requires 4 parameters per atom. An ellipsoid needs 6 parameters thus an anisotropic atom requires 9 parameters. This is an example of an anisotropic atom Final stages of refinement. There are many was in which a structure can be “improved”. The two most important considerations are addition of hydrogen atoms and anisotropic refinement of the non-hydrogen atoms. Addition of hydrogen atoms – Hydrogen atoms have only 1 electron and are often not seen in difference maps. It is best to include them at calculated positions. This is easy to do and it will improve the “R factor”. R Factor – The R factors used are Rw and wR2. Rw should be < 8% and wR2 should be <15%. The lower the better. Rs are of the form Sum[(I0-Ic)/Io] pma 2019
Problems with single crystal X-ray Crystallography Locating Hydrogen atoms - Hydrogen atoms make extremely small contributions and for this reason X-ray crystallography is not a good technique for accurately locating hydrogen atom positions. If the location of hydrogen atoms is of specific interest (e.g. in the study of hydride structures and hydrogen bonding interactions) use has got to be made of the much more expensive and less available technique of neutron diffraction. The theory of neutron diffraction is very similar to that for X-ray diffraction but an essential difference is that hydrogen atoms scatter neutrons as effectively as many other atoms and for this reason they can be located with good accuracy in the structure determination. The Need for Single Crystals - In order to carry out a detailed X-ray structure determination, it is essential to have a crystal of the material in question. Many compounds cannot be crystallised and thus are not amenable to diffraction studies. There are also commercially important materials such as glasses and many ceramics which owe their unique properties to their amorphous nature. Being amorphous (no long range order), the structures of these materials cannot be investigated in detail by diffraction techniques. Low Temperature Structure Determination – When X-ray data are collected at low temperature (<-150 ºC) thermal ellipsoids are smaller and better defined. N.B. bond lengths show very little variation with temperature. pma 2019
Using CheckCif to examine the quality of a structure • The IUCrCheckCif service should be used to check the quality of crystal structures • It is available online at https://checkcif.iucr.org/ • Problems/Alerts are ranked A, B, C etc. • A and B alerts should be examined and fixed if possible. • C alerts often indicate ways in which structures can be improved. pma 2019
A Really Good Structure. In this structure the diff map peaks are drawn as lime green spheres and you can see that they are all "bond blobs" caused by the effects of chemical bonding which slightly distorts the spherical atoms. This is the limit of what can be achieved using the spherical atom model. pma 2019
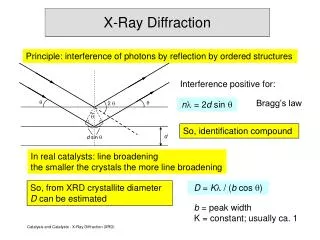
X-Ray Powder Diffraction (XRPD) A crystalline powder sample will diffract X-rays but since the orientations of the individual crystals are random the data set produced is a plot of intensity v.s. diffraction angle or Bragg angle . Here the sample is sitting on a flat plate and the plate is turned about the centre of the diffractometer at half the rate through which the detector moves. This is the /2 or Bragg scan method. Notice the plot contains 2 on the X-axis and X-ray intensity on the y-axis. pma 2019
Uses of X-ray Powder Diffraction In general, powder diffraction data are unsuitable for solving crystal structures. Some advances have recently been made using the Rietveld method. However this is far from trivial and it works best in relatively simple cases. It is very difficult to be sure that the unit cell is correct as the reflections overlap and are difficult to resolve from one another. There may also be problems with preferred orientation of crystallites. Important advantages and uses of powder diffraction: 1. The need to grow crystals is eliminated. 2. A powder diffraction pattern can be recorded very rapidly and the technique is non-destructive. 3. With special equipment very small samples may be used (1-2mg.) 4. A powder diffraction pattern may be used as a fingerprint. It is often superior to an infrared spectrum in this respect. 5. It can be used for the qualitative, and often the quantitative, determination of the crystalline components of a powder mixture. 6. Powder diffractometry provides an easy and fast method for the detection of crystal polymorphs. Powder patterns are provided when a drug is being registered with the FDA. (Polymorphs are different crystal forms of the same substance.) pma 2019
Preferred Orientation Effects in XRPD It is possible to calculate the theoretical diffraction pattern if the crystal structure is known. Nifedipine Calculated pattern Observed pattern There are no preferred orientation effects here as all reflections have their expected intensity. pma 2019
002 004 There is clear preferred orientation here. The 002 is the flat face exposed when the needles lie down on a flat plate. XRPD of Benzoic acid Calculated pattern Observed pattern pma 2019
Some points relating to preferred orientation effects. • Preferred orientation effects are often observed for needles and plates. • Preferred orientation effects can be reduced by sample rotation and sample grinding. • When an indexed calculated pattern is compared to that of a sample showing preferred orientation it may be possible to to index the faces of plate like crystals. • Deviations from calculated patterns can be used to monitor the crystal morphology of production batches. pma 2019
Calculations using X-ray powder diffraction patterns. For an orthogonal system (a = b = = 90°) the relationship between interplanar spacing (d) and the unit cell parameters is given by the expression: This is the expression for an orthorhombic crystal. For the tetragonal system it reduces to and, for the cubic system, it further reduces to pma 2019
In the CsCl structure both ions have coordination numbers of 8 and the structure is a simple primitive one with no centring. Formula Cs at centre = 1 8 x 1/8Cl = 1 = CsCl Important Cubic Lattice Types Two of the most important cubic lattice types are the NaCl type and the CsCl type. NaCl crystallizes in the Space Group Fm-3m Stoichiometry (formula) from the Unit Cell Site Na+ Cl- Central 0 1 Face 6/2 0 Edge 0 12/4 Corner 8/8 0 Total 4 4 pma 2019
Cubic close packed spheres The unit cell of a cubic close packed Metal has a face cantered or F type lattice The formula of the unit cell is: 6 x ½ + 8 x 1/8 = 4 pma 2019
The Bragg equation may be rearranged (if n=1) from to If the value of 1/(dh,k,l)2 in the cubic system equation above is inserted into this form of the Bragg equation you have Since in any specific case a and l are constant and if l2/4a2 = A pma 2019
Analysis Cubic XRPD patterns Example 1 Aluminium powder gives a diffraction pattern that yields the following eight largest d-spacings: 2.338, 2.024, 1.431, 1.221, 1.169, 1.0124, 0.9289 and 0.9055 Å. Aluminium has a cubic close packed structure and its atomic weight is 26.98 and l = 1.5405 A . Problem - Index the diffraction data and calculate the density of aluminium. can be used to obtain sin, The Bragg equation, The ccp lattice is an F type lattice and the only reflections observed are those with all even or all odd indices. that are allowed Thus the only values of sin2 in are 3A, 4A , 8A, 11A, 12A,16A and 19A for the first eight reflections. pma 2019
Insert the values into a table and compute sin and sin2. Since the lowest value of sin2 is 3A and the next is 4A the first Entry in the Calc. sin2 column is (0.10854/3)*4 etc. The reflections have now been indexed. pma 2019
Calculation of a For the first reflection (for which h2 + k2 + l2 = 3) sin2 = 3A = 3 ( l2 / 4a2 ) a2 = 3l2 / 4sin2 a = 4.04946 Å = 4.04946 x 10-8 cm. Calculation of the density of aluminium a3 = 66.40356 Å3 = 66.40356 x 10-24 cm3. If the density of aluminium is r (g. cm.-3), the mass of the unit cell is x 66.40356 x 10-24 g. The unit cell of aluminium contains 4 atoms. The weight of one aluminium atom is 26.98/(6.022 x 1023) = 4.48024 x 10-23 and the weight of four atoms (the content of the unit cell) is 179.209 x 10-24. r x 66.40356 x 10-24 = 179.209 x 10-24 p = 2.6988 g.cm-3. pma 2019
Example 2 The X-ray powder diffraction pattern of AgCl obtained using radiation of wavelength 1.54Å is shown below. The peaks are labelled with 2θ values Calculate the following: On the basis that the structure is cubic and of either the NaCl or CsCl type 1. Index the first six reflections. , 2. Calculate the unit cell parameter, 3. Calculate the density of AgCl. (Assume the following atomic weights: Ag, 107.868; Cl, 35.453; and Avogadro’s number is 6.022 x 1023) pma 2019
Since values are available sin2 values can be calculated and inserted in a table. From Sin2 = A(h2 + k2 + l2) the possible values are: 1. for a face centred lattice 3A, 4A , 8A, 11A, 12A and 16A 2. for a primitive lattice 1A, 2A, 3A, 4A, 5A and 6A The second option is not possible as the first 2 are not in the ratio of 1:2. To test the first option, divide the first by 3 and multiply the result by 4, 8 etc. pma 2019
Density of AgCl Since sin2 = l2(h2 + k2 + l2)/4a2 a2 = (1.54)2.(16)/4(0.3083) using the largest (most accurate) 2 a2 = 30.7692 a = 5.547Ǻ (1Ǻ=10-8 cm) Formula wt. of unit cell = 4AgCl = 573.284g This is the weight of 4 moles of AgCl. The weight of 4 molecules is 573.284 / (6.02 x 1023) Density = 573.284 / (6.02 x 1023)(5.547 x 10-8)3 A is in Ǻ thus the answer should be multiplied by 1 / 10-24 Density= 5.580 g/cm3 pma 2019