Download

1 / 52
530 likes | 651 Views
Sulfonamides and Antimicrobial Antifolates (Step 1).

E N D
Ehrlich was the father of anti-microbial chemotherapy and also the father of immunology. He came up with the idea of a ‘magic bullet’ that had a different toxicity for the parasite than for the host. His principles for discovering anti-infectives included the notion that synthetic compounds can cure infections, you need to systematically explore structures and activities, you needed standard infection models with reliable endpoints, you strive to raise the therapeutic index (LD50/ED50), and that drug resistance may arise in the pathogen against a class of chemicals
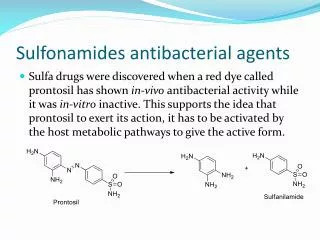
He worked with arsenic and azo dyes, discovering the uses of compounds like Salvarsan (agent 606 for syphilis), and trypan red/blue or afridol for trypanosomiasis. • Gerhard Domagk thought dyes would potentially make good drugs, since they bound proteins in wool. He contributed to the discovery of Prontosil, the first highly effective and reliably nontoxic antibacterial agent. Prontosil is a sulfonamide (-SO2NH2).
It was determined that the Prontosil is metabolized to Sulfanilamide (SA), its active form. SA was active both in vitro and in vivo, and is produced from Prontosil by metabolic activation in the tissues. Regardless of whether SA or Prontosil is administered, patients excrete SA. • Sulfonamides structurally resemble PABA (p-aminobenzoic acid). Their mechanism involves the inhibition of folic acid synthesis; they competitively inhibit dihydropteroate synthetase by competing with PABA: • Pteridine + PO4 (pterin pyrophosphokinase)→ __ + PABA (dihydropteroate synthetase)→ __ + glutamate → dihydrofolate (dihydrofolate reductase)→ folate.
The most common way resistance arises is by mutation in the dihydropteroate synthetase enzyme to escape the effects of sulfonamides. Resistance may also arise from reduced permeability to sulfonamides, increased expression of dihydropteroate synthetase, or increases in PABA to outcompete the sulfonamide. • Sulfonamides are selectively toxic against microbes because they synthesize folic acid, whereas we cannot. We don’t have the enzyme dihydropteroate synthetase. Furthermore, microbes are impermeable to folic acid, so even if it were present in the environment, they still wouldn’t take it up. We actively transport folic acid.
Lots of derivatives have been synthesized, but only a dozen or so have reached clinical use, and only three are now used. Sulfanilamide is insoluble in the urine, and may result in stones. Substituting at various locations on the molecule may influence the antimicrobial spectrum, therapeutic index, solubility, metabolic pattern, half life, permeability, distribution, absorption, etc.
The sulfonamides presently in clinical use are Sulfisoxazole (Gantrisin – most widely used), Sulfamethoxazole (Ganatol), and Sulformethoxine (Sulfadoxine, Fanasil – 150 hour half life, used sparingly). • Toxicity: Adverse reactions requiring cessation of therapy are rare (3%), though in AIDS patients they are quite common. They may include fever, rash, joint pain, and lymphadenopathy. Very few of these reactions are dose-dependent. Long acting sulfonamides may cause serious Stevens-Johnson syndrome, so should only be administered for good reasons.
Sulfonamides should NEVER be given to newborns. Very high death rates were observed in premature infants treated with sulfonamides. Deaths were associated with kernicterus, yellow staining of the basal ganglia due to bilirubin deposits in the brain. Sulfonamides bind to serum albumin and displace bilirubin from being bound to the albumin. This leads to more free bilirubin, and more gets into the brain and binds there. Total serum bilirubin levels will be LOW, since a lot of it gets displaced and the unbound form equilibrates with other body compartments.
Sulfonamides are cheap, orally bioavailable, and continue to be used. They may be used for prophylaxis of simple urinary tract infections from gram (-) bacteria. They are the drug of choice for Nocardia, a genus of gram (+) bacteria. They may be used for other things too. • Related compounds are used for leprosy (mycobacterium leprae – Dapsone) and tuberculosis (p-aminosalcyclic acid – PAS). • Many drugs are structurally related to sulfonamides. Some for goiters, anti-diabetic agents, gout, acetazolamide (diuretic), etc.
Elion and Hitchings developed trimethoprim, as well as mercaptopurine, pyrimethamine, acyclovir (antiviral), and allopurinol (gout). These two studied purine and pyrimidine synthesis, and spent a lot of time studying diaminopyrimidines. Aminopterin and methotrexate are folic acid analogs, and they are diaminopyrimidines.
Elion and Hitchings synthesized trimethoprim, which was a highly effective antibacterial agent. It inhibits folate biosynthesis at DHFR. Resistance to it may result from reduced susceptibility to the drug, or less commonly over-production of DHFR. Both humans and bacteria have DHFR, but trimethoprim is selective for the bacterial DHFR. The human and bacterial versions are different, even though they carry out the same reaction. • Trimethoprim toxicity is pretty rare. It may be rash/nausea/vomiting. Or you can get folate deficiency in nutritionally deprived patients, resulting in anemia, thrombocytopenia, and neutropenia.
They then tried combination chemotherapy with both sulfonamides and trimethoprim, and found a synergistic result. This combination of trimethoprim and sulfamethoxazole was sold as Bactrim/Co-trimoxazole. It had a broader spectrum of activity than either drug alone, was less likely to have bacteria develop resistance, less toxicity due to smaller doses of each drug, and is bacteriocidal rather than bacteriostatic. It’s useful for UTIs, otitis media, shigellosis, chronic bronchitis, pneumocystis carinii, salmonella typhi, and MRSA.
Introduction to Antibacterials • An antibiotic is an antibacterial substance produced by a microorganism. Basically drugs that kill or stop the growth of bugs. • Individualizing therapy requires that you integrate clinical medicine, microbiology and pharmacology related to the particular infectious disease. Patients with the same underlying problem may be very different and have different needs based on the clinical syndrome, organism, appropriate drug class and individual agent that best balances cost, convenience, side effects, etc.
You begin with broad, empiric therapy. To do this, consider the probability of different organisms given the clinical syndrome, patient, and geographic factors (community vs. hospital [nosocomial] acquired). Hospital acquired stuff is usually more resistant and lethal. You should consider the lethality of possible, but less probable, pathogens as well as local antibiotic sensitivity patterns. • Eventually, you want to work toward a definitive therapy that is narrower. You’d like to choose the best drug for the organism, but also for the patient’s situation. This isn’t as dependent on probabilities. • You want a narrow spectrum of coverage to avoid unnecessary resistance, unnecessary adverse side effects, for simplicity of management and to reduce cost of treatment.
There are lots of obstacles to choosing the right antibiotic. They include syndromes caused by multiple agents and agents that cause multiple syndromes, variations with population/geography/age, agents may be unidentifiable by clinical findings, or simply not knowing the cause of a syndrome. There are also so many organisms and so many drugs, it’s tough to put it all together. • We should try to know, for a class of pathogens, the antibiotics that incrementally cover them. And groups of organisms with similar antibiotic sensitivities. Basically, know the grid.
More things to consider when choosing the right drug are: mechanism of action, selective toxicity, spectrum of activity, bacteriocidal/bacteriostatic, combination antibiotics, synergy vs. antagonism between drugs. • The selective toxicity of an antibiotic is the relationship between the toxicity in bacteria in relation to toxicity in the host. Selective toxicity = bacterial toxicity / host toxicity. The therapeutic window = host efficacy / host toxicity. • Selective toxicity is often due to the different biochemistry between eukaryotes and prokaryotes. There are differences in cell wall (cell wall active drugs), DNA gyrase (quinolones), RNA polymerase (rifampin), ribosomes (ribosomal inhibitors), folate synthesis/transport (sulfonamides), and dihydrofolate reductase (trimethoprim).
Even though human and bacterial receptors may be only distantly related, drugs that act on bacteria still produce a lot of side effects in us. • The adverse effects are often unrelated to antimicrobial effects: • Β-lactams: CNS, bleeding, nephrotoxicity, allergy • Vancomycin: histamine release • Aminoglycosides: nephrotoxicity, ototoxicity, neuromuscular • Tetracyclines: discoloration of teeth and bones • Erythromycin: hepatitis, GI
Clindamycin: pseudomembranous colitis • Chloramphenicol: aplastic anemia, bone marrow • Quinolones: CNS toxicity, arthropathy, QTc prologation • Sulfonamides: hemolytic anemia • Trimethoprim: folate antagonism.
Bacteriocidal drugs kill the microorganisms. A bacteriostatic drug alone (with immune dysfunction or in a sanctuary site like the CNS) will prevent proliferation. A bacteriostatic drug with good immune system and penetration will kill the bugs as well. Generally, bacteriocidal drugs include penicllins, cephalosporins, quinolones, aminoglycosides, and trimethoprim/sulfamethoxazole. Bacteriostatic drugs include tetracycline, macrolides, chloramphenicol, clindamycin, sulfonamides, and trimethoprim.
The distinction is variable, with some drugs being cidal for some microorganisms and static for others: • Macrolides: static against atypicals and GNR, cidal against S. pyogenes and S. pneumoniae. • Chloramphenicol: static against S. aureus, cidal against S. pneumoniae. • Linezolid: static against Staphylococci and Enterococci, cidal against Streptococci. • There are a lot of situations where either a bacteriocidal or static agent may be appropriate.
It may be good to use combinations of antibiotics for mixed infections, preventing resistance (for TB since it’s particularly prone to developing resistance), initial empiric therapy for a serious infection, or for a synergistic effect (more than additive). Synergy is pretty uncommon, an example might be penicillin and aminoglycosides. • Antibiotic tissue penetration depends on all the usual stuff (concentration, size, protein binding, transport, etc), but also inflammation. Inflammation may make a membrane more permeable for some drugs. But for β-lactams, pumps that remove the drug from the CNS are less active in inflammation. So these drugs accumulate in the CNS during inflammation.
The extravascular characteristics may also alter drug concentrations. If there’s a large reservoir, big changes in serum concentration may only manifest as small changes in the tissues. Or, if a tissue has pumps getting rid of the drug, extravascular concentrations of the drug may be quite low regardless of serum concentrations
Loading doses may be used to rapidly achieve a target concentration. • Various drugs have different distributions to the CNS, urine and other compartments. In the CNS, penicillin has medium penetration (1% normally, but 5% with inflammation). • Barriers to drug penetration include the outer membrane of gram (-) bacteria, cell walls, periplasmic beta-lactamases, the cytoplasmic membrane, and cytoplasmic pumps.
The time course of a drug often doesn’t parallel the time course of its effect. Some drugs’ effects are concentration dependent (quinolone, aminoglycosides, tetracycline), but others are not and may be time dependent (beta-lactam, vancomycin, oxazolidinone, macrolides, clindamycin). For time dependent ones, as long as the concentration is >4xMIC, increasing the concentration gives no greater effect. • Many also exhibit a Post Antibiotic Effect (PAE), in which cidal/static properties continue after a short exposure to the drug. Beta lactam antibiotics exhibit PAE against gram (+) organisms, but not gram (-) ones. Aminoglycosides and quinolones show PAE vs. gram (-) organisms.
Cell Wall Inhibitors Gram (+) bacteria have a surface of thick peptidoglycan, with some teichoic acid, overlaying the plasma membrane. Gram (-) bacteria have an outer membrane with LPS (may act as a barrier for some drugs), a periplasmic space with a single layer of peptidoglycan, and an inner membrane.
The peptidoglycan of the cell wall is made of chains of alternating NAG and NAM, which are cross-linked by peptide bridges. Gram (-) rods have a modified amino acid, DAP, followed by d-Ala d-Ala. Gram (+) cocci have a lysine with a pentaGlycine followed by d-Ala d-Ala. Cross linking occurs at these residues just before the d-Ala d-Ala. • Transglycosidase joins NAGs and NAMs, and transpeptidase cross-links strands of NAG/NAM. Several other enzymes act to lay down peptidoglycan. In the process of producing peptidoglycan, you have to break down and displace some of the existing layer. So there’s continually new wall being synthesized and broken apart.
The d-Ala d-Ala sequence is targeted by transpeptidase for cross-link formation. D-Ala isn’t present in vertebrates. The terminal d-Ala is cleaved, and the remaining one forms a covalent complex with the enzyme. This covalent bond to the d-Ala is then replaced with the peptide bond cross-link to another chain of peptidoglycan. • The beta-lactam ring of penicillin and other beta-lactam antibiotics holds these molecules in a conformation that is homologous to the d-Ala d-Ala on which transpeptidase acts. Penicillin inhibits transpeptidation by forming a covalent complex with the enzyme. Autolysins (peptidoglycan hydrolases) continue to dissolve the cell wall, and since it’s not being synthesized the wall becomes depleted. In a way, the bacteria’s own enzymes destroy it.
The enzymes that bind penicillin and are involved in peptidoglycan synthesis and remodeling are called penicillin binding proteins (PBPs - transpeptidase, transglycosylase, etc). They may have various specific functions, like elongating the cell, maintaining shape, forming septa, etc. Their sensitivities to beta-lactam antibiotics also varies, and not all PBP inhibition is lethal. • Beta lactam antibiotics can cause significant morphological changes in the microbes they affect, due to alteration of cell wall parameters. • These antibiotics have selective toxicity (toxic for bacteria more than us) because bacteria have cell walls, d-Ala d-Ala, and transpeptidease whereas we do not.
Penicillin G: • Acts on gram (+) Strep as well as oral (above-the-belt) anaerobes. When it first came out it was effective against most microorganisms, but resistance has changed that. • It is acid unstable and erratically absorbed, so you can’t take it orally. • It’s distributed into 50% of the body weight, and has poor penetration into the CSF due to an organic anion transport (OAT) pump. Inflammation inhibits the pump and increases penetration into the CSF.
Only 10% of it gets metabolized, but some of the products may be allergenic. • It has a very short half life of 45 minutes, and is excreted by the kidneys through glomerular filtration and secretion by an organic anion pump. Probenecid inhibits this pump and lowers the excretion rate. • Beta-lactam antibiotics are bactericidal, have time-dependent killing (the more time you spend over the MIC, the more you kill), and have a post antibiotic effect (PAE) for gram (+) bacteria. The PAE is suppression of bacterial growth that persists after short exposure of organisms to antimicrobial agents. It allows dosing to be less frequent that suggested by the half life.
Resistance to penicillin may arise by alterations in penicillin binding protein sites, reduced permeability of the drug, or production of beta-lactamase to degrade the antibiotic by opening its beta-lactam ring. Beta-lactamase is plasmid mediated, and this enzyme is excreted. It is constitutively expressed and present in the periplasm in many gram (-) bacteria. To counter this, we have developed beta-lactamase inhibitors: Clavulanate, Sulbactam, Tazobactam. • Clavulanate competes with penicillin for access to the beta-lactamase active site, and the beta-lactam ring of clavulanate is opened instead of penicillin’s.
Augmentin = clavulanate + amoxicillin. Much more potent that amoxicillin alone. • Beta-lactamase inhibitors have limitations. These include activity against only class A serine hydroxylases, though tazobactam is active against other classes. Also, the inhibitor is degraded after binding. Some resistance has emerged, and multiple classes of beta-lactamase may be produced by one organism (or multiple organisms present may result in the presence of several beta-lactamases). Finally, clavulanate induces beta-lactamase expression, which is counterproductive. Future development will be focusing on expanding the classes of enzymes on which the drugs will work, as well as increasing membrane permeability.
Adverse effects of penicillin include • Hypersensitivity (any type may occur, but type I is rare/life-threatening). The most common toxic side effect is GI symptoms with oral drugs. Ticarcillin may result in a sodium overload. Rarely, you may see bone marrow depression, hepatitis, platelet aggregation, or seizures. • If someone has a history of type I anaphylactic reaction to penicillin, they’ll give a skin test before administering a beta lactamase antibiotic. If the skin test is positive, 50-70% of patients will experience anaphylaxis with penicillin. A positive skin test will also be cross-reactive against cephalosporin 30% of the time. If the test is negative, only 1-3% will have minor cutaneous reactions which aren’t a big deal.
Drawbacks of penicillin include its short half-life, instability in gastric acid, inactivation by beta-lactamase, poor treatment of gram (-) bacteria, and its allergenicity. • You can modify penicillin side chains to try to increase its stability against beta-lactamases, increase its binding to PBPs, alter its pharmacokinetics, make it more acid stable, or make it more permeable to gram (-) bacteria.
Derivatives of penicillin have been made to increase its spectrum against gram (-)s (amoxicillin, ampicillin, ticarcillin and pipercillin), improve its pharmacokinetic parameters (penicillin V, penicillin G benzathine/procaine), and be resistant to penicillinases (methicillin, oxacillin, cloxacillin, dicloxacillin). • Longer acting penicillins like procaine penicillin, benzathine penicillin (very slow!) and orally available penicillin V made the half life longer. Benzathine penicillin is good against syphilis, because you need a really low dose for a long time, and it gives just that.
Aminopenicillins: Ampicillin acts on some gram (-) bacteria including H. influenza, it is 50% orally bioavailable, and it doesn’t cause allergy (though it does cause diarrhea and rash). Combining these with a beta-lactamase inhibitor can be very effective against beta-lactamase producing Staph, Neisseria, and H. influenza. Amoxicillin + clavulanate = Augmentin (oral).
Acts on gram (+) Pneumococcus, Streptococcus, and Enterococcus. Acts on gram (-) H. influenzae, E. coli, Proteus mirabilis, and Salmonella. • Augmentin increases spectrum to include beta-lactamase producing S. aureus, H influenzae, M catarrhalis and many gram (-) rods. • Amoxicillin is 100% orally bioavailable. Combining these with a beta-lactamase inhibitor can be very effective against beta-lactamase producing Staph, Neisseria, and H. influenza. Ampicillin +sulbactam = Unasyn (IV).
Antipseudomonal aminoacylpenicillins: piperacillin, piperacillin/tazobactam, mezlocillin, azlocillin. o Piperacillin covers what ampicillin does, but also more nosocomial (class II gram (-) rods), Pseudomonas aeruginosa, Bacteroides fragilis. Tazobactam extends coverage to beta-lactamase producing strands of staphylococci and many GNR, though it’s no better against pseudomonas than piperacillin alone since pseudomonas doesn’t use beta-lactamase. The combo is used for serious gram (-) infections.
Ticarcillin shows time dependent killing. In this situation, a continuous infusion could be used to avoid falling below a minimum or rising above a maximum desired concentration. Continuous infusion actually showed better results than intermittent dosing too. • Gram (+) bacteria produce “penicillinase” which is basically the same as beta-lactamase from gram (-) bacteria. Penicillinase resistant penicillins include methicillin, oxacillin, cloxacillin, dicloxacillin, and nafcillin. MRSA isn’t resistant to the antibiotics because of beta-lactamase, rather due to altered PBPs, so it’s also resistant to all beta-lactam antibiotics.
These penicillinase resistant penicillins are active against penicillinase producing Staphylococci, Pneumococci, and group A Streptococci. • Staph aureus and epidermidis (MRSA and MRSE) are resistant to these. They are also resistant to cephalosporins and imipenem. • Dorothy Hodgkin figured out the structure of penicillin.
Pharmacokinetics of penicillins: Generally (except ceftriaxone) they have short half lives, renal clearance predominates, and have variable protein binding. Their distribution is poor to the eye, brain, CSF and prostate, and inflammation usually enhances penetration. • Dosing: despite half life of .5-1.5 hours, you can dose every 4 hours because of the PAE. The exceptions are for meningitis you want every 2 hours, amoxicillin only needs to be given every 8 hours, and benzathine and procaine penicillin have very long half lives.
Penicillins are one class of beta-lactam antibiotics. Another is the cephalosporins. They also have a beta-lactam ring, they have a broad spectrum of activity [gram (+/-) and pseudomonas], they are resistant to beta-lactamases, and are safer than penicillins. There are four ‘generations’ of cephalosporins based on their spectrum of activity. Generations progressively increase in their coverage of gram (-) bacteria.
1st: cephazolin, cephalothin, cephalexin - good for strep and staph aureus. • 2nd: cefotetan – better gram (-) coverage, a little worse gram (+). Surgeons use a lot, good for Bacteroides fragilis and interabdominal infections. • 3rd: ceftriaxone, ceftazidime – even better gram (-) coverage. • 3rd/4th: cefepime – more resistant to some beta-lactamases.
Cephalosporins show PAE against gram (+) organisms. They are characterized by time dependent killing, so continuous infusion is better. • Adverse effects of cephalosporins include hypersensitivity and anaphylaxis (same as penicillin, but not as common). • Unique cephalosporins: Ceftriaxone (3rd gen) can be given once daily, whereas most are 3x daily. Cefotetan (2nd gen) is particularly good at battling anaerobes. Cefepime (4th gen) is particularly potent against pseudomonas.
A third group of beta-lactam antibiotics are the carbapenems. These include meropenem and imipenem/Cilastatin. Pseudomonas have a protein in their outer membrane just for the drugs in this class, and allows the drugs to get in. In some cases, these pores are the only way for drugs to get into these gram (-) bacteria. • They have a broad spectrum. They kill gram (+)s except Enterococcus faecium and MRSA. They kill gram (-)s including pseudomonas. They also kill anaerobes. And, so far resistance to them is rare.
They have a PAE for gram (-) and (+) bacteria. • Carbapenems are associated with seizures, though the incidence isn’t well defined. • Meropenem has a 1 hour half life, but can be given every 8 hours due to PAE. It is also metabolized in the kidneys, in addition to being filtered and secreted there. Proximal tubular dehydropeptidase I hydrolyzes the meropenem in the urine. The metabolite is a renal tubular toxin, though, so it’s often administered with Cilastatin to inhibit the renal dehydropeptidase I. This allows more of the meropenem itself to be excreted. • Aztreonam has only gram (-) coverage with no cross-allergenicity with penicillins, so you can use it without fear of anaphylaxis. It’s also a non-aminoglycoside.
Vancomycin is a huge molecule, and it only works on gram (+) bacteria. It H-bonds with the d-Ala d-Ala and pentaglycine of gram (+) peptidoglycan so that transpeptidases can’t link them together. Some Enterococci are vancomycin resistant. Resistance may arise by changing d-Ala d-Ala to d-Ala d-Lac. • Vancomycin enters the CSF poorly with uninflamed meninges, but improves with inflammation. There is little metabolism of it. It is excreted renally. Half life is usually around 6 hours. Its killing is time dependent. • Vancomycin toxicity includes allergenicity (drug fever, rash, not anaphylaxis), phlebitis, and red man flushing with a rapid IV dose. No good evidence for oto or nephrotoxicity.
Ribosomal Inhibitors The prokaryotic ribosome is a complex of protein and RNA. All together it is 70S, and it includes a 50S subunit and a 30S subunit. tRNA may bind to an A-site, P-site, or E-site. There is a peptidyl transferase cavity where the main reaction occurs; lots of drugs act here. There is also a peptide exit tunnel where the elongating chain sits.
Polypeptide elongation: empty tRNA is in the E-site, tRNA-polypeptide is in the P-site and exit tunnel. A tRNA/AA half-enters the A-site (A/T), then fully enters (A/A). The polypeptide is moved to the tRNA in the A-site. The empty tRNA leaves the E site, and the remaining tRNAs shift (P/E and A/P). Then they fully shift to the E-site (E/E) and P-site (P/P). • Gentamicin (aminoglycoside): bacteriocidal, acts at the 30S A-site. Blocks initiation, terminates, and causes misreads. • Tetracycline: bacteriostatic, acts at the 30S A-site. Blocks tRNA binding (A/T to A/A)
Chloramphenicol: bacteriostatic, acts at the 50S A-site. Blocks peptidyl transferase. • Clindamycin: bacteriostatic, acts at the 50S A and P sites. Blocks peptidyl transferase. • Linezolid (Oxazolidinone): bacteriostatic, acts at the 50S P-site. Blocks fMet rTNA initiation complex. • Erythromycin (Macrolide): bacteriostatic, acts at the 50S exit site. Blocks the peptide exit tunnel.