Download

1 / 38
590 likes | 1.16k Views
Exome Sequencing as Molecular Diagnostic Tool of Mendelian Diseases. BIOS 6660 Hung-Chun (James) Yu Shaikh Lab 04/28/2014. Human Genetic Diseases. Penetrance vs F requency. Kaiser J. S cience (2012) 338:1016-1017. Human Genetic Diseases. Complex Disorder Polygenic, many genes.

E N D
Exome Sequencing as Molecular Diagnostic Tool of Mendelian Diseases BIOS 6660 Hung-Chun (James) Yu Shaikh Lab 04/28/2014
Human Genetic Diseases • Penetrance vs Frequency Kaiser J. Science (2012) 338:1016-1017.
Human Genetic Diseases • Complex Disorder • Polygenic, many genes. • Low penetrance/effect size. • Multifactorial, environmental, dietary. • Examples: heart disease, diabetes, obesity, autism, etc. • Mendelian Disorder • Monogenicor polygenic. • Full or high penetrance/effect size. • Examples: sickle cell anemia and cystic fibrosis.
Complex Diseases • Multiple causes, and polygenic. • Multiple genetics factors with low penetrance individually. Coronary artery disease Coriell Institute for Medical Research. https://cpmc1.coriell.org/genetic-education/diagnosis-versus-increased-risk
Mendelian Diseases Veltman J.A. et al. Nat. Rev. Genet. (2012) 13:565-575.
Mendelian Diseases • Dominant Inheritance U.S. National Library of Medicine. http://ghr.nlm.nih.gov/
Mendelian Diseases • Recessive Inheritance U.S. National Library of Medicine. http://ghr.nlm.nih.gov/
Exome Sequencing Bamshad, MJ., et al. Nat. Rev. Genet. (2011) 12:745-755.
Exome Sequencing • ~40Mb (coding) or 60Mb (coding + UTRs)
Mendelian Diseases Identified by Exome Sequencing • Timeline Gilissen C. et al., Genome Biol. (2011) 12:228.
Mendelian Diseases Identified by Exome Sequencing • By mid-2012, ~100 genes identified. • By mid-2013, >150 genes identified. Rabbani, B., et al. (2012) J. Hum. Genet. 57:621-632.
Types of Variation • What kind of variation/mutation can be detected by Exome Sequencing? • SNV (single nucleotide variation) • Small InDel, (insertion/deletion of <25bp) • Large InDel, CNV (copy number variation) • Possible, but not reliable. • Aneuploidy • Same as CNV • Translocation • Possible, but not reliable. Limited. • Complex rearrangement • Not likely.
Exome Variants • SNV (single nucleotide variation) • Synonymous: (1) Silent. • Nonsynonymous: (1) Missense. (2) Nonsense. (3) Stop-loss. (4) Start-gain. (5) Start-loss. (6) Splice-site. http://upload.wikimedia.org/wikipedia/commons/6/69/Point_mutations-en.png http://www.web-books.com/MoBio/Free/Ch5A4.htm
Exome Variants • Small InDel (insertion/deletion <25bp) • Frameshift • In-frame NHGRI Digital Media Database (DMD), http://www.genome.gov/dmd/
Variant and Population Frequency • Novel/Private variant • Never been reported before. • Rare variant • Minor allele freq. (MAF) < 1%. • Polymorphic variant • MAF > 1% (0.01) or 5% (0.05). • Databases • dbSNP (NCBI): http://www.ncbi.nlm.nih.gov/SNP/ • 1000 Genomes: http://www.1000genomes.org/ • ESP (NHLBI): http://evs.gs.washington.edu/EVS/
Exome Variants • How to analyze enormous amount of variants in any given exome? Private/Novel ~100 - 300 Protein altering ~4,000 - 15,000 Coding + splice-site ~10,000 - 30,000 All ~20,000 - 200,000 GilissenC. et al. Eur. J. Hum. Genet. (2012) 20:490-497.
Exome Variants Bamshad, MJ., et al. Nat. Rev. Genet. (2011) 12:745-755.
Exome Analysis Strategies Male Female Affected Heterozygous carrier Sex-linked heterozygous carrier Mating Consanguineous mating GilissenC. et al., Eur. J. Hum. Genet. (2012) 20:490-497.
Exome Analysis Strategies • Linkage • Large family with multiple affected individuals • Pathogenic variant co-segregate with disorder. • Homozygosity • Affected patients from consanguine parents. • Homozygous mutation within a homozygous stretch in the genome. • Ideal for recessive disorders
Exome Analysis Strategies • Candidate genes • Biased approach • Require current biological knowledge • Good for screening or clinical diagnosis of known disorders. • Overlap • Require multiple unrelated individuals with identical disorders. • Monogenic disorders
Exome Analysis Strategies • De novo • Sporadic mutation • Germline mutation during meiosis • Dominant inheritance *
Exome Analysis Strategies • Double-hit • Unaffected parents are heterozygous carries • Parental sequence info is very helpful • Recessive inheritance. Homozygous Compound Heterozygous * * * * * * # #
Trio-based Exome sequencing • Family trio • Unaffected parents and an affected patient. • Why we use trio? What can be tested using trio? Advantages? • Economical, efficient, single case required.
Trio-based Exome sequencing • X-linked dominant • De novo • X-linked recessive • Hemizygous in male • Autosomal dominant • De novo • Autosomal recessive • Compound heterozygous • Homozygous Male Female Affected * Heterozygous carrier Sex-linked heterozygous carrier X X Y X * Y X
Trio-based Exomesequencing • Candidate Genes/Variants • Protein altering variants • Rare or novel variants • Variants that fit each inheritance model
Case 1 • Clinical information The patient was a 7-month-old boy when first evaluated. He was diagnosed with BPES by a pediatric ophthalmologist. In addition to blepharophimosis, ptosis, and epicanthus inversus normally associated with BPES, he had cryptorchidism, right hydrocele, wide-spaced nipples, and slight 2–3 syndactylyof toes. Clinical testing demonstrated a normal karyotype (46,XY), and normal FISH studies for 22q11.2 deletion, Cri-du-Chat (5p deletion) syndrome. Thyroid function was normal. Further, normal 7-dehydrocholesterol level was used to rule out Smith–Lemli–Opitzsyndrome. Sanger sequencing and highresolutionCNV analysis with Affymetrix SNP 500K arrays did not identify a FOXL2 mutation.
Case 1 • A-D: 2-month old. note blepharophimosis, ptosis, epicanthus inversus (A), posteriorly angulated ears with thickened superior helix and prominent antihelix (B), and slight 2–3 syndactyly of toes in addition to overlapping toes (C, D) • E-F: 3.5-year old. Following oculoplastic surgery to correct ptosis; note right-sided preauricular ear pit (F, indicated by arrow). • G-I: 12-year old. Note the recurrence of ptosis (L>R), arched eyebrows, abnormal ears, thin upper lip vermilion, small pointed chin, downsloping shoulders, and wide-spaced and low-set nipples.
Case 2 • Clinical information The proband is a nine year old girl who presented with microcephaly, unilateral retinal coloboma, bilateral optic nerve hypoplasia, nystagmus, seizures, gastroesophagealreflux, and developmental delay including not yet saying specific words (at 29 months old). On exam, she has microcephaly with a normal height, a down-turned upper lip, and fingertip pads. A karyotype and CGH analysis have been normal. Kabuki (KMT2D and KDM6A) and Angelman (UBE3A and MECP2) syndromes were suspected in this patient.
Case 3 • Clinical information Case 3 was the result of a non-consanguineous union and he presented to care at four months of age with a seizure disorder, hypotonia and developmental delay. The patient underwent a left parietal craniotomy and partial resection of the frontal cortex without complete resolution of the seizure disorder. Initial laboratory studies included an elevated homocysteineand methylmalonic acid and a normal vitamin B12 level. Complementation analysis of the patient’s cell line placed the patient into the cblC class. Sequencing and deletion/duplication analysis (microarray) the MMACHC gene was negative in both skin fibroblasts and peripheral blood.
Case 3 • Feature Combined methylmalonicaciduria and homocystinuria. Severe developmental delay, infantile spasms, gyralcortical malformation, microcephaly, chorea, undescended testes, megacolon
Case 3 • Monster Max http://www.maxwatson.org/ • Patient's older sister as a summer student in Shaikh Lab
Data for Case Study • 3 trios • A total of 3 families/cases. • Each family/case includes unaffected parents and an affected patient. • VCF files • Familial variants calls in VCF format, mapped to human GRCh37/hg19. • 2x90bp paired-end reads, with ~50X coverage • “Mini” Exome • 100 genes with/without known disorder association. • Validated causative genes, plus randomly selected genes.
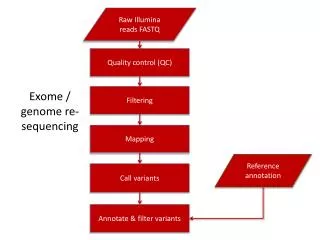
Exome NGS Workflow FASTQ 2x90bp BCF Filter based on Phredscore, mapping quality, read depth, etc. SAM Filter unpaired, unmapped reads VCF ? BAM Filter PCR duplicates artifact BWA (Burrows-Wheeler Aligner) SAMtools
VCF Format • VCF (Variant Call Format) http://www.1000genomes.org/wiki/Analysis/Variant%20Call%20Format/vcf-variant-call-format-version-41 ## Meta-information lines FILTER, INFO, FORMAT # Header line
VCF Format • INFO • AA : ancestral allele • AC : allele count in genotypes, for each ALT allele, in the same order as listed • AF : allele frequency for each ALT allele in the same order as listed: use this when estimated from primary data, not called genotypes • AN : total number of alleles in called genotypes • BQ : RMS base quality at this position • CIGAR : cigar string describing how to align an alternate allele to the reference allele • DB : dbSNP membership • DP : combined depth across samples, e.g. DP=154 • END : end position of the variant described in this record (for use with symbolic alleles) • H2 : membership in hapmap2 • H3 : membership in hapmap3 • MQ : RMS mapping quality, e.g. MQ=52 • MQ0 : Number of MAPQ == 0 reads covering this record • NS : Number of samples with data • SB : strand bias at this position • SOMATIC : indicates that the record is a somatic mutation, for cancer genomics • VALIDATED : validated by follow-up experiment • 1000G : membership in 1000 Genomes
VCF Format • FORMAT • GT: Genoetype. 0/0: Homozygous normal 0/1: Heterozygous variant 1/1: Homozygous variant • PL: the Phred-scaled genotype likelihoods (>0). 0/0 0/1 1/1 174 ,0 ,178 • GQ : Genotype quality (1-99)